Thanatophore Dysplasie (TD)
Die Thanatophore Dysplasie (TD) ist eine autosomal-dominante Skelettdysplasie, die durch einen schweren dysproportionalen Kleinwuchs, schmalen Thorax und Makrocephalus gekennzeichnet ist. Sie wird in zwei Subtypen unterteilt: TD1 mit einer “Telefonhörerform” des Femurs und TD2 mit einer geraden Femurform und möglicher Kleeblattdeformation des Schädels. Die Erkrankung resultiert aus pathogenen Varianten im FGFR3-Gen, wobei verschiedene Varianten unterschiedliche Krankheitsausprägungen hervorrufen können.
Wissenschaftlicher Hintergrund
Die Thanatophore Dysplasie (TD) ist eine Erkrankung mit autosomal-dominantem Erbgang (bzw. wird durch dominante Neumutationen verursacht) und einer Inzidenz von 1 : 20.000 – 40.000. Die TD ist damit eine der häufigeren Formen letaler Skelettdysplasien. Klinisch ist die Erkrankung durch schweren dysproportionierten Kleinwuchs mit rhizomel verkürzten Extremitäten, einem sehr schmalen Thorax sowie Makrocephalus (evtl. Kleeblattschädel) charakterisiert. Betroffene Kinder sterben meist in den ersten Lebensstunden, jedoch gibt es Patienten, die mehrere Jahre alt werden.
Die Thanatophore Dysplasie wird in zwei Subtypen unterteilt:
- Typ 1 (TD1): kurze, gebogene Oberschenkelknochen (“Telefonhörerform” des Femurs)
- Typ 2 (TD2): gerade, relativ lange Femurform, mehr oder weniger stark ausgeprägte Kleeblattdeformation des Schädels
Die Erkrankung wird durch pathogene Varianten im FGFR3-Gen (Fibroblasten-Wachstums-Faktor-Rezeptor 3) verursacht. Eine TD1 kann z.B. durch Aminosäuresubstitutionen in der extrazellulären oder intrazellulären Domäne des Proteins entstehen. Beispiele für die extrazelluläre Domäne sind die beiden häufigen Varianten R248C und Y373C. Es wurden aber auch das Stopp-Codon betreffende Varianten berichtet, die zu einer Verlängerung des Proteins führen. Homozygotie für eine mit Achondroplasie assoziierte Veränderung führt ebenfalls zum Phänotyp einer TD.
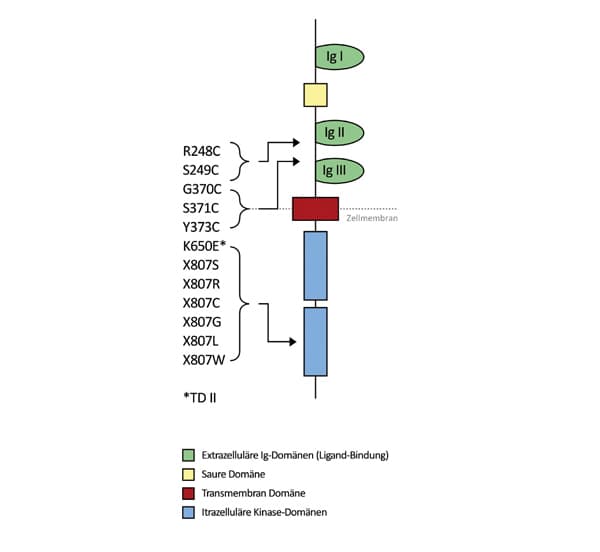
Abb.: Schematische Darstellung der Domänen des FGFR3-Proteins, der Verankerung des Proteins in der Zellmembran und die Lokalisation der Mutationen im Protein (modifiziert nach Hilbert et al. 1998, Monatsschr Kinderheilkd 146:687)
Die einzige relevante Veränderung bei der TD2 (FGFR3-K650E) ist in nahezu allen TD2-Fällen nachweisbar. Im selben Codon des FGFR3-Gens können allerdings auch Varianten auftreten, die mit einer weniger schwerwiegenden Erkrankungsform, der SADDAN-Dysplasie, oder mit der mildesten Ausprägung der FGFR3-Erkrankungen, der Hypochondroplasie, assoziiert sind. Dies lässt darauf schließen, dass es in Abhängigkeit vom Aminosäureaustausch zu einer unterschiedlich starken Veränderung der Tyrosinkinaseaktivität des Rezeptors kommt (Genotyp-Phänotyp-Beziehung).
Erkrankung | ICD—10 | Gen | OMIM—G |
| Thanatophore Dysplasie Typ 1 | Q77.1 | FGFR3 | 134934 |
Wainwright 2016, S Afr Med J 106:S50 / Xue et al. 2014, Mol Genet Genomic Med 2:497 / Foldynova-Trantirkova et al. 2012, Hum Mutat 33:29 / Martinez-Frias et al. 2009, Am J Med Genet Part A 152A:245 / Lievens et al. 2004, J Biol Chem 279:43254 / Zabel 2004, medgen 16:8 / Van Esch et al. 2004, Genet Counsel 15:375 / Wilkin 2001 in: The Metabolic & Molecular Basis of Inherited Disease 5379 / Vajo et al. 2000, Endocr Rev 21:23 / Perez-Castro et al. 1997, Genomics 41:10 / Tavormina et al. 1995, Nature Genet. 9:321
letzte Aktualisierung: 23.4.2024