Hämochromatose, hereditäre
Die Hämochromatose ist eine autosomal-rezessive Erkrankung, die durch eine Störung des Eisenstoffwechsels verursacht wird und zu einer Eisenakkumulation in verschiedenen Organen führt. Die erbliche Form der Hämochromatose wird in etwa 80% der Fälle durch Homozygotie für den Aminosäureaustausch p.(Cys282Tyr) (C282Y; rs1800562) im HFE-Gen verursacht. Die Penetranz ist unvollständig, so dass nur 15-25% der homozygoten Merkmalsträger an einer klinisch manifesten Hämochromatose erkranken. Heterozygote Merkmalsträger haben kein erhöhtes Erkrankungsrisiko. Der Aminosäureaustausch p.(His63Asp) (H63D; rs1799945) ist ein Risikofaktor für eine leichte Eisenakkumulation, insbesondere in Kombination mit anderen Risikofaktoren. Bei Patienten mit unauffälliger Basisdiagnostik und mittels Leberbiopsie, Magnetresonanz-Bildgebung oder quantitativer Phlebotomie nachgewiesener schwerwiegender Eisenüberladung, bei denen sonstige Lebererkrankungen oder hämatologische Erkrankungenausgeschlossen wurden, ist eine erweiterte molekulargenetische Diagnostik indiziert. Dabei besteht die Möglichkeit des Nachweises seltener Varianten in den Genen HFE, HJV (alias HFE2), HAMP, TFR2, SLC40A1 und BMP6.
Wissenschaftlicher Hintergrund
Bei der Hämochromatose handelt es sich um eine sogenannte late-onset-Erkrankung, die auf einer erworbenen (sekundären) oder angeborenen (hereditären) Störung des Eisenstoffwechsels beruht. Das Manifestationsalter liegt bei Männern in der Regel zwischen der 4. bis 6. Lebensdekade und bei Frauen ab etwa dem 50. Lebensjahr. Durch eine erhöhte Eisenresorption im Dünndarm kommt es zu einer Eisenakkumulation in verschiedenen Organen, insbesondere in der Leber. Bei frühzeitiger Diagnose ist die Erkrankung gut therapierbar, unbehandelt führt sie häufig zu Leberzirrhose, Lebertumoren, Diabetes mellitus oder Herzrhythmusstörungen. Bei der erblichen Form (hereditäre Hämochromatose, HH) handelt es sich um eine Erkrankung, die in ca. 80% der Fälle durch Homozygotie für den Aminosäureaustausch p.(Cys282Tyr) (C282Y; rs1800562) im autosomal-rezessiv vererbten HFE-Gen verursacht wird. Die Häufigkeit dieses Genotyps liegt im mitteleuropäischen Raum zwischen 1:200 und 1:400. Die Penetranz ist unvollständig, so dass nur 15-25% der homozygoten Merkmalsträger an einer klinisch manifesten Hämochromatose erkranken. Der Nachweis dieses Genotyps bei symptomatischen Patienten kann als Bestätigung für eine HH gesehen werden. Heterozygote Merkmalsträger (5-10% der Bevölkerung) haben kein erhöhtes Erkrankungsrisiko. Der ebenfalls bekannte Aminosäureaustausch p.(His63Asp) (H63D; rs1799945) ist nicht mit hereditärer Hämochromatose assoziiert, aber in homozygoter Form ein Risikofaktor für eine leichte Eisenakkumulation, insbesondere in Kombination mit anderen Risikofaktoren wie z.B. Alkohol, Übergewicht oder bestimmten Stoffwechselerkrankungen.
Bei Patienten mit klinischem Verdacht auf eine hereditäre Hämochromatose aufgrund einer Transferrinsättigung von >45% und einem Serumferritinwert von >200 µg/l wird eine genetische Untersuchung des HFE-Gens (Basisdiagnostik Genotypisierung) empfohlen.
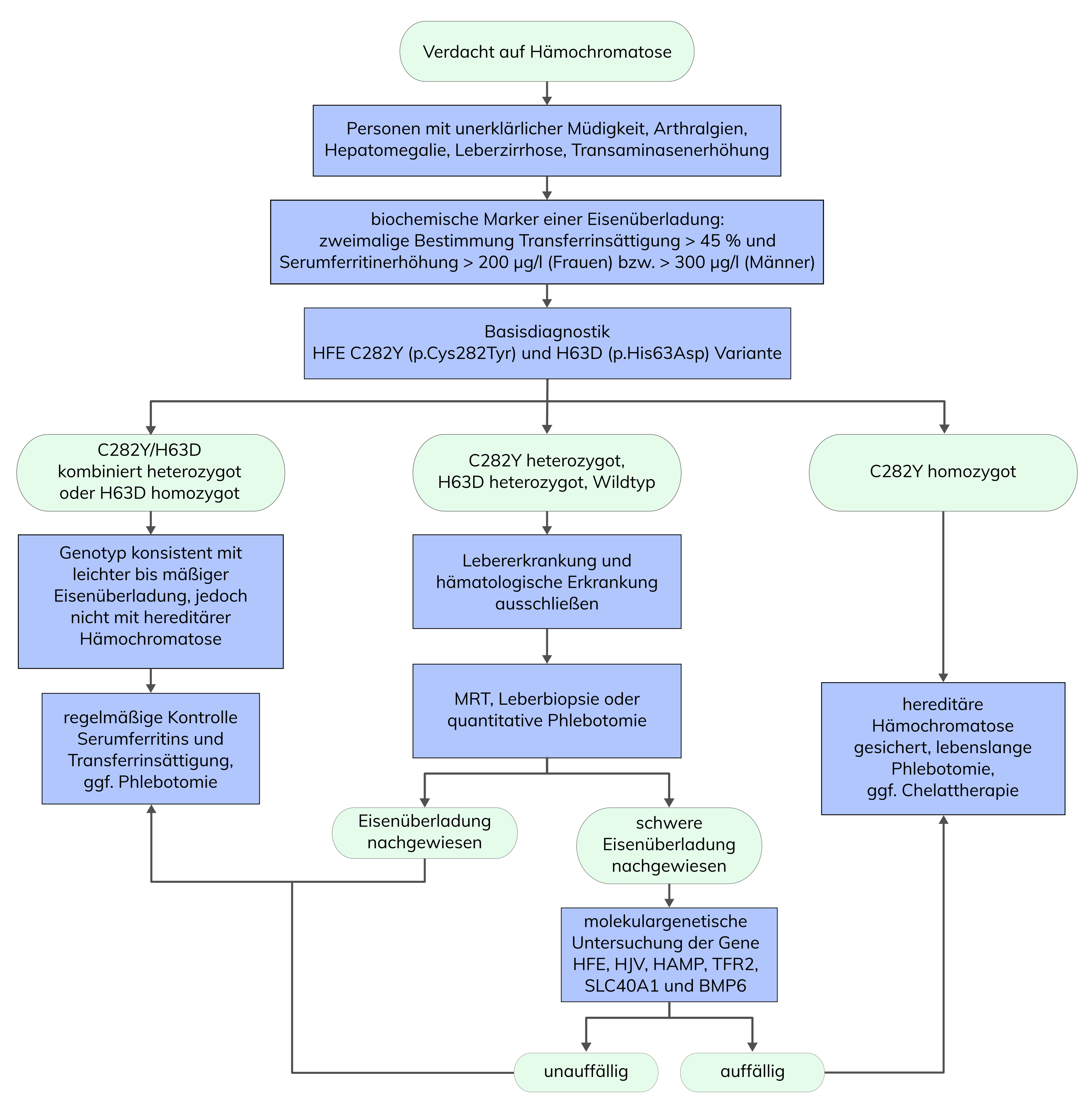
Bei Patienten mit unauffälliger Basisdiagnostik und mittels Leberbiopsie, Magnetresonanz-Bildgebung oder quantitativer Phlebotomie nachgewiesener schwerwiegender Eisenüberladung, bei denen sonstige Lebererkrankungen oder hämatologische Erkrankungenausgeschlossen wurden, ist eine erweiterte molekulargenetische Diagnostik indiziert. Dabei besteht die Möglichkeit des Nachweises seltener Varianten in den Genen HFE, HJV (alias HFE2), HAMP, TFR2, SLC40A1 und BMP6.
Die juvenile Hämochromatose tritt typischerweise im ersten bis dritten Lebensjahrzehnt auf und ist gekennzeichnet durch das Auftreten einer schweren Eisenüberladung (Serumferritinkonzentration von 1.000 bis 7.000 µg/l) und einer sehr hohen Transferrin-Sättigung, die oft 100% erreicht. Männer und Frauen sind gleichermaßen betroffen. Prominente klinische Merkmale umfassen u.a. hypogonadotropen Hypogonadismus, Kardiomyopathie und Leberzirrhose. Die Haupttodesursache ist eine Herzerkrankung. Wenn die juvenile Hämochromatose früh genug erkannt wird und regelmäßig ein Aderlass durchgeführt wird, werden Morbidität und Mortalität stark reduziert. Veränderungen in Genen, die eine juvenile Hämochromatose verursachen, betreffen das HJV-Gen, das für Hämojuvelin codiert und mehr als 90% der Erkrankungen verursacht und das HAMP-Gen, das für Hepcidin codiert und weniger als 10% der Erkrankungen verursacht. Beide Gene werden autosomal-rezessiv vererbt.
Bei Patienten mit einer nachgewiesenen hereditären Hämochromatose ist ein lebenslanger, regelmäßiger Aderlass die Therapie der Wahl, die bei frühem Beginn zu einer normalen Lebenserwartung führt. Kann ein Aderlass nicht durchgeführt werden, kann eine Therapie mit Chelaten in Betracht gezogen werden. Zudem wird eine gesunde, ausgewogene Ernährung empfohlen.
Hämochromatose Typ | Gen (Vererbung) | Funktion | Symptome / weitere Manifestationen | Eintritt | Pathogenese | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hämochromatose Typ 1 klass. Hämochromatose | HFE (AR) homeostatic iron regulator | bindet an Transferrinrezeptor und reduziert die Affinität für Eisen-beladenes Transferrin | erhöhte TS und SF, Eisenüberladung in Leber | adult | ineffektive/reduzierte Hepcidin vermittelte Ferroportinregulation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hämochromatose Typ 3 | TFR2 (AR) Transferrinrezeptor 2 | vermittelt die zelluläre Aufnahme von Transferrin- gebundenem Eisen | erhöhte TS und SF, Eisen in Hepatozyten | adult | ineffektive/reduzierte Hepcidin vermittelte Ferroportinregulation | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Hämochromatose Typ 4a nicht-klassische Ferroportin-Krankheit | SLC40A1 (AD) Ferroportin | beteiligt am Eisenexport aus der Zwölffingerdarmepithelzelle; vermittelt den Eisen-Efflux in Gegenwart einer Ferroxidase (Hephaestin und / oder Ceruloplasmin) | erhöhte TS und SF, Eisen in Hepatozyten | adult | Ferroportin Gewinn durch übermäßigem zellulären Eisenexport | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
weitere Inhalte anzeigen | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Barton & Edwards 2018, GeneReviews® [Internet] / De Gobbi & Roetto 2018, GeneReviews® [Internet] / Rihl 2017, Akt Rheumatol 42:529 / Wang et al. 2017, Int J Hematol 105:521 / Daher et al. 2016, Gastroenterology 150:672 / Porto et al. 2016, Eur J Hum Genet 24:479 / Goldberg 2011, GeneReviews® [Internet] / Sang et al. 2011, Cell 145:513 / Wolf & Hildebrandt 2011, Ped Nephrol 26:181 / Clark et al. 2010, Clin Biochem Rev 31:3
letzte Aktualisierung: 5.11.2023