Hämoglobinopathien - Übersicht
Hämoglobinopathien sind eine Gruppe genetischer Erkrankungen, die durch eine gestörte Bildung von Hämoglobin gekennzeichnet sind und sich in zwei Hauptgruppen einteilen lassen: Thalassämien und strukturelle Hämoglobinvarianten. Die klinisch bedeutsamen α- und β-Thalassämien resultieren aus einer quantitativen Störung der Globinketten-Synthese, die zu einer leichten bis schweren Anämie führen kann. Strukturelle Defekte der Globinketten charakterisieren die anomalen Hämoglobine wie HbS, HbC und HbE. Die Diagnose von Hämoglobinopathien erfordert die Abgleichung hämatologischer Befunde mit molekulargenetischen Ergebnissen, besonders im Kontext eines Kinderwunsches oder bei bekannter Familienanamnese.
Wissenschaftlicher Hintergrund
Die Hämoglobinopathien umfassen eine Gruppe von genetischen Erkrankungen, denen eine gestörte Bildung von Hämoglobin (Hb) zugrunde liegt. Sie lassen sich in zwei Hauptgruppen unterteilen: Thalassämien und anomale Hämoglobine (strukturelle Hämoglobinvarianten).
Thalassämien
α- und β-Thalassämie
Bei den Thalassämien sind die α- und β-Thalassämie aufgrund ihrer Häufigkeit und der Schwere der Erkrankung von besonderer klinischer Bedeutung. Sie beruhen auf einer quantitativen Störung der jeweiligen Globinketten-Synthese.
δβ-Thalassämie & Hereditäre Persistenz fetalen Hämoglobins (HPFH)
Der Großteil der δβ-Thalassämien und HPFH basiert auf großen Deletionen im β-Globin-Genkomplex. Beide Formen zeichnen sich durch eine signifikante Erhöhung des fetalen Hämoglobins HbF aus, das bei Erwachsenen normaler Weise nur in sehr geringen Mengen vorliegt (<1%). Die phänotypischen Unterschiede der einzelnen Formen sind wie folgt aufgeführt:
Anomale Hämoglobine
Im Gegensatz zu den Thalassämien beruhen die anomalen Hämoglobine hauptsächlich auf einem strukturellen Defekt der betroffenen Globinkette. Zu den häufigsten anomalen Hämoglobinen zählen:
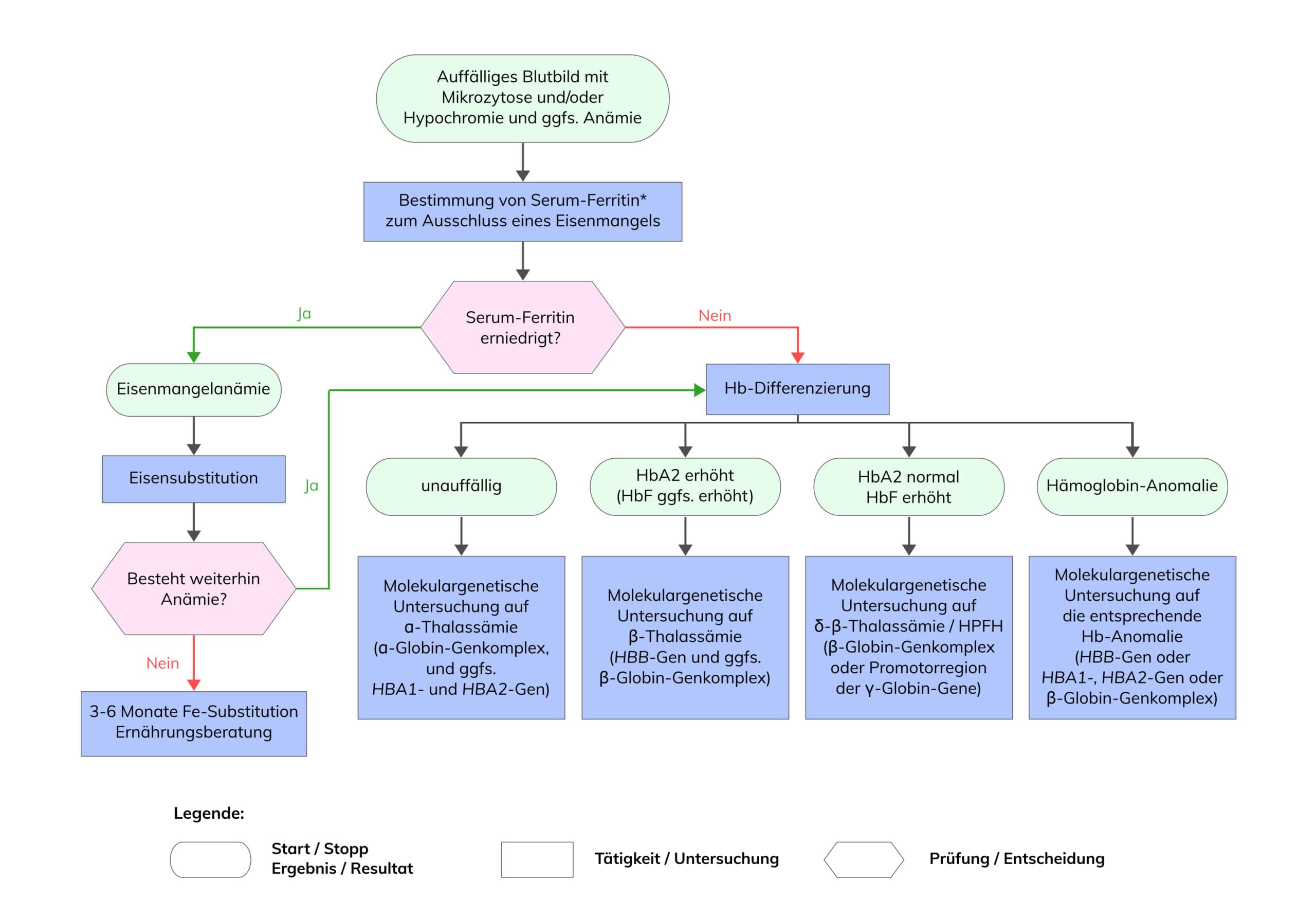
Bei der Diagnostik von Hämoglobinopathien ist zu beachten, dass diese in den verschiedensten Kombinationen auftreten können. Daher sollten die hämatologischen Befunde (, , ) immer mit den Ergebnissen aus der Molekulargenetik abgeglichen werden.
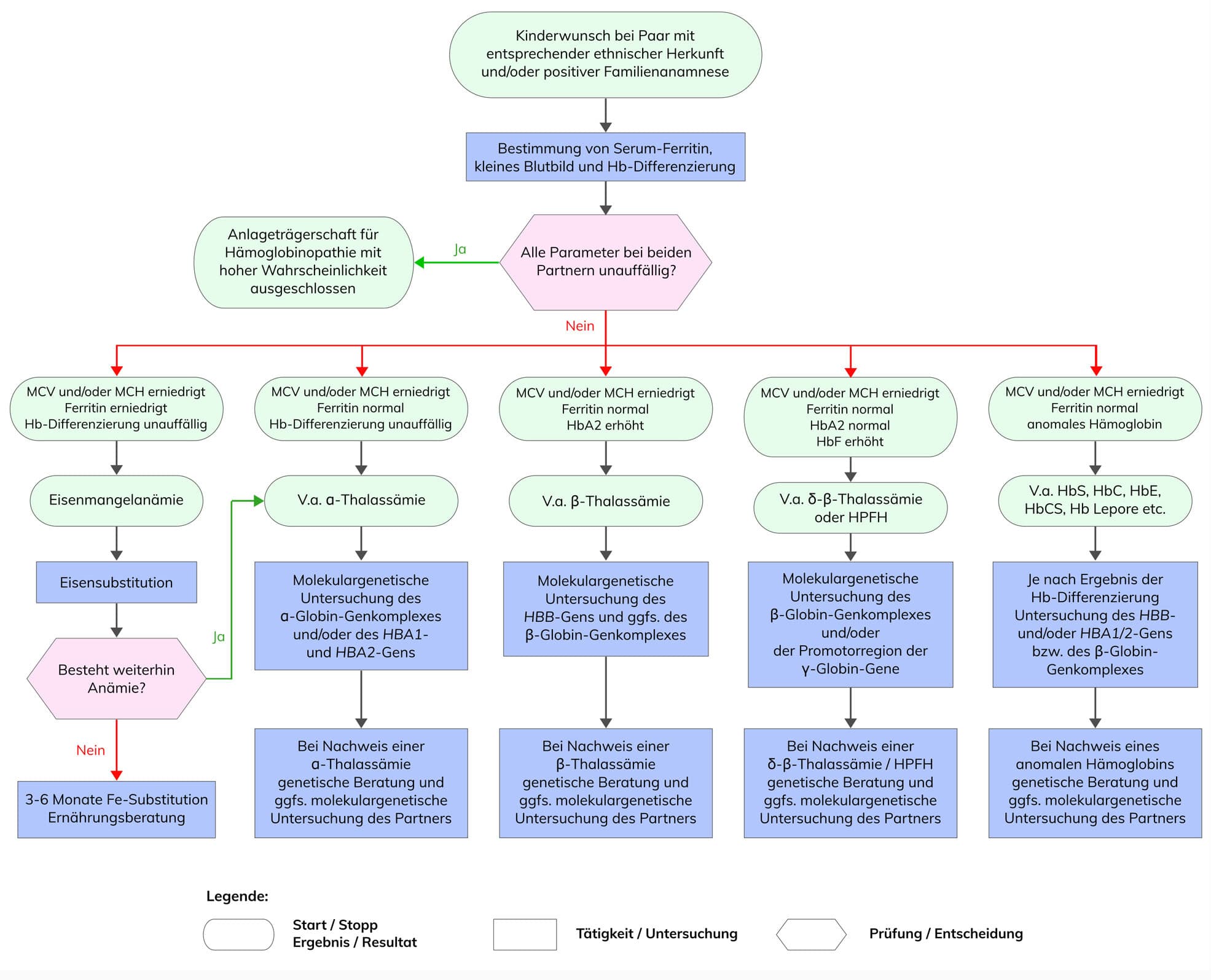
Häufig stellt sich die Frage nach der Anlageträgerschaft einer Hämoglobinopathie erst im Rahmen eines Kinderwunsches, entweder aufgrund einer positiven Familienanamnese oder durch die ethnische Herkunft. Bei einer bereits bestehenden Schwangerschaft ist eine zügige Diagnostik wichtig, um so früh wie möglich das Risiko einer schweren Form einer Hämoglobinopathie bei Nachkommen abschätzen zu können.
Die detaillierte diagnostische Vorgehensweise zur allgemeinen Diagnostik von Hämoglobinopathien und zur Vorgehensweise bei Paaren mit Kinderwunsch ist in den folgenden Flussdiagrammen dargestellt.