Essentielle Thrombozythämie (ET)
Die Essentielle Thrombozythämie (ET) ist eine myeloproliferative Neoplasie, die durch eine übermäßige Produktion von Thrombozyten (>450×10^9/L), eine Anhäufung großer Megakaryozyten im Knochenmark und eine mögliche Entwicklung zu Myelofibrose oder akuter myeloischer Leukämie charakterisiert ist. Für die Diagnose sind Hauptkriterien wie Thrombozytenzahl, Knochenmarkbiopsie, Abwesenheit anderer myeloischer Erkrankungen und das Vorhandensein spezifischer Genvarianten (JAK2, CALR oder MPL) entscheidend. Verschiedene Genvarianten, insbesondere in JAK2, MPL und CALR, sind mit unterschiedlichen klinischen Merkmalen und Risiken für thrombotische Komplikationen verbunden, während zytogenetische Veränderungen bei etwa 5-10% der Patienten zum Diagnosezeitpunkt beobachtet werden.
Wissenschaftlicher Hintergrund
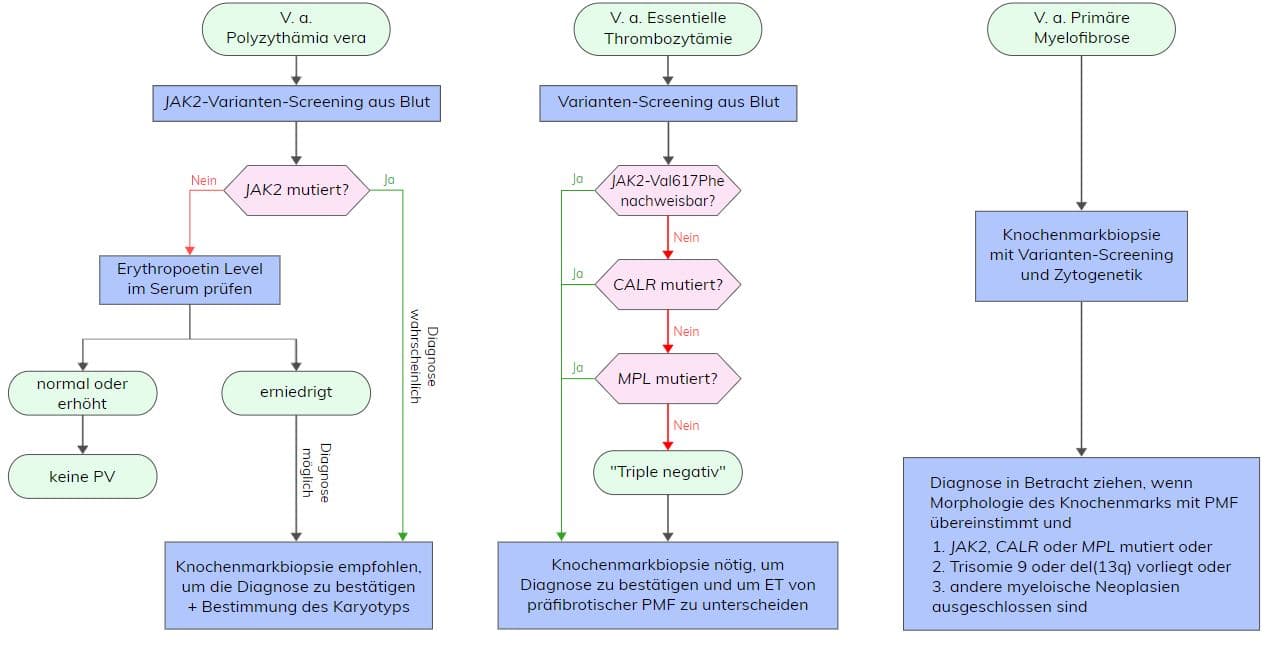
Die Essentielle Thrombozythämie (ET) gehört zu den myeloproliferativen Neoplasien (MPN) und ist eine hämatopoetische Stammzellerkrankung charakterisiert durch eine klonale oder polyklonale Expansion der Thrombozytopoese und Thrombozytose (>450×109/L) im peripheren Blut, eine erhöhte Anzahl großer, stark gelappter Megakaryozyten im Knochenmark und dem Auftreten von Thrombozytose und/oder Haemorrhagie. Die Inzidenz wird mit 0,2 – 2,3/100.000/Jahr angegeben. Die Erkrankung tritt meist im Alter von 50-60 Jahren auf und hat noch einen zweiten Alterspeak um die 30 Jahre. Etwa <4% der Patienten zeigen einen Übergang in eine sekundäre Myelofibrose und <2% in eine . Für die Diagnose einer ET müssen nach WHO alle vier Hauptkriterien oder die ersten drei Hauptkriterien und ein Nebenkriterium erfüllt sein.
Hauptkriterien:
- Thrombozytenzahl >450×109/L
- Knochenmarkbiopsie: Proliferation hauptsächlich der Megakaryozytenlinie
mit erhöhter Anzahl vergrößerter reifer Megakaryozyten mit stark gelappten
Zellkernen; keine signifikante Erhöhung oder Linksverschiebung der neutrophilen
Granulopoese oder Erythropoese und sehr selten geringe (Grad 1) Faserbildung - keine zutreffenden WHO-Kriterien für BCR-ABL1+ CML, PV, PMF, MDS oder
andere myeloide Neoplasie - Anwesenheit einer JAK2, CALR oder MPL Variante
Nebenkriterien: Anwesenheit eines klonalen Markers oder Fehlen von Beweisen für eine reaktive Thrombozytose
Zu den molekulargenetisch-diagnostischen Kriterien der ET gehört eine Variante in Exon 14 des JAK2-Gens (Val617Phe), die in etwa 50-60% der ET-Patienten nachgewiesen werden kann. Das entstehende Protein verfügt über eine gesteigerte Tyrosinkinase-Aktivität. Die Janus-Kinase 2 ist bei Gesunden essentiell für die Blutbildung. Über sie führen Zytokine wie Erythropoetin, Thrombopoetin oder G-CSF zu einer Aktivierung der STAT-Moleküle in der Zelle und zu einer Proliferation hämatopoetischer Zellen. Eine Variante in JAK2 führt zu einer Liganden-unabhängigen, konstitutiven Signaltransduktion. Hierdurch kommt es zu einer unkontrollierten Proliferation von Blutzellen. Generell zeigen Patienten mit dieser Variante meist ein höheres Alter, höhere Hämoglobin- und Leukozytenwerte und geringere Thrombozytenwerte.
In ca. 3-5% der ET-Patienten können Varianten in Exon 10 des MPL-Gens, die hauptsächlich die Aminosäure Trp515 betreffen, nachgewiesen werden. Das MPL-Gen kodiert für den Thrombopoietin-Rezeptor, welcher vor allem für die Proliferation von Megakaryozyten wichtig ist und über Thrombopoietin (TPO) aktiviert wird. Dadurch kommt es zu einer Stimulation des JAK/STAT-Signalweges. Die häufigsten Varianten an MPL-Trp515 führen zu einem Aminosäureaustausch von Tryptophan nach Leucin (Trp515Leu), Lysin (Trp515Lys) oder Alanin (Trp515Ala) und somit zu einer konstitutiven Aktivierung des JAK-STAT-Signalweges. Eine weitere Variante (MPL-Ser505Asn) wurde neben sporadischen Fällen auch im Zusammenhang mit familiärer ET beschrieben. Es sind auch andere, seltenere MPL-Varianten (Ala506Thr, Ala519Thr, Leu510Pro) beschrieben, deren klinische Relevanz bislang aber unklar ist. ET-Patienten mit MPL-Variante zeigen meist höhere Thrombozyten- und Erythropoetinwerte, geringeres Hämoglobin und Knochenmarkzellularität sowie fortgeschrittenes Alter. Verglichen mit JAK2 Val617Phe-mutierten ET-Patienten besteht bei Patienten mit MPL-Variante ein höheres Risiko für thrombotische Komplikationen.
Weitere ca. 30% der ET-Patienten tragen Varianten in Exon 9 des CALR-Gens. Dabei handelt es sich um verschiedene somatische Deletions- bzw. Insertionsvarianten, die zu einem Frameshift und somit zu einem spezifischen, alternativen Leserahmen führen. Man unterscheidet dabei vorrangig Typ 1- (52 bp-Deletion, p.(Leu367fs*46)) bzw. Typ 1-ähnliche Varianten und Typ 2- (5 bp-Insertion, p.(Lys385fs*47)) bzw. Typ 2-ähnliche Varianten in CALR, wobei in ET Typ 2- bzw. Typ 2-ähnliche Varianten mit signifikant höheren Thrombozytenwerten assoziiert sind. CALR codiert für ein Ca2+-bindendes Chaperon des endoplasmatischen Retikulums. ET-Patienten mit CALR-Varianten sind im Vergleich zu JAK2-Val617Phe- bzw. MPL-mutationspositiven ET-Patienten jünger, haben eine deutlich erhöhte Thrombozytenzahl, niedrigeres Hämoglobin, verminderte Leukozytenzahlen sowie ein geringeres Thromboserisiko.
In JAK2-, CALR– oder MPL-mutierten Patienten kann die Varianten-Allel-Frequenz (VAF) hilfreich zur Klärung der spezifischen Diagnose sein. So ist z.B. eine 40% JAK2-VAF unüblich für eine ET und deutet eher auf eine maskierte PV oder präfibrotische/frühe PMF hin.
ET-Patienten, die keine Variante in JAK2, MPL oder CALR aufweisen, werden als „triple-negativ“ bezeichnet. In Einzelfällen kann bei diesen Patienten eine SH2B3-Variante nachgewiesen werden. SH2B3 spielt eine entscheidene Rolle in der Hämatopoese durch negative Regulation der JAK2 Aktivierung. Die häufigsten Varianten finden sich in Exon 2 des SH2B3-Gens, gefolgt von Exon 8.
Etwa 53% der Patienten mit ET zeigen weitere Varianten wie TET2 (16%), ASXL1 (11%), DNMT3A (6%) und SF3B1 (5%). Risikofaktoren für Gesamtüberleben, Myelofibrose-freies oder Leukämie-freies Überleben sind Varianten in SH2B3, SF3B1, U2AF1, TP53, IDH2 und EZH2, von denen mindestens eine in 15% der ET-Patienten detektiert wird.
Zum Zeitpunkt der Diagnose sind zytogenetische Veränderungen bei ca. 5-10% der Patienten mit ET detektiertbar. Zu den beobachteten, aber nicht beständigen Veränderungen gehören Trisomie 8, Deletionen im langen Arm von Chr. 20 und Deletionen im kurzen Arm von Chr. 9.