Ehlers-Danlos-Syndrom (EDS)
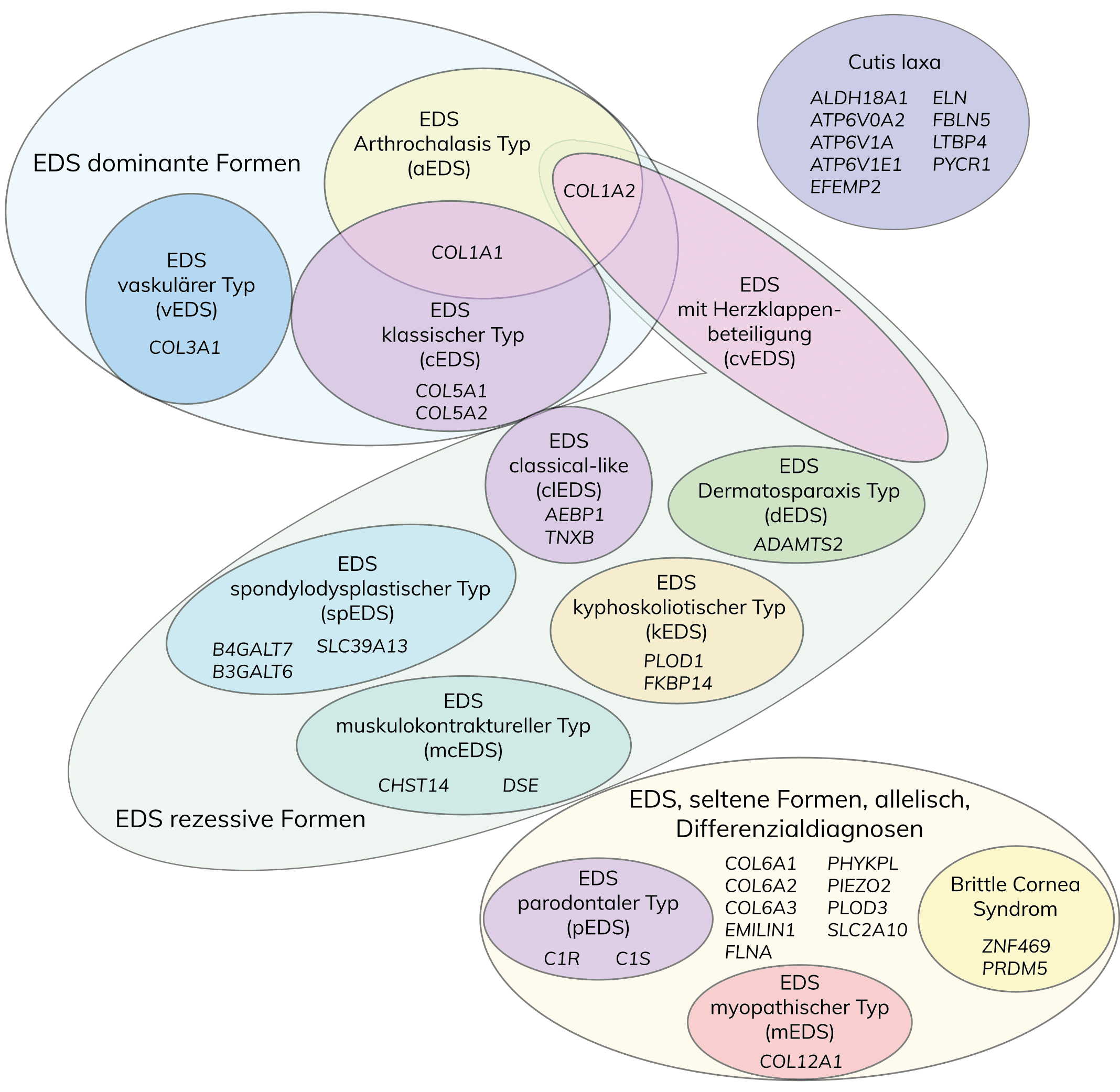
Das Ehlers-Danlos-Syndrom (EDS) ist eine Gruppe genetischer Erkrankungen des Bindegewebes, gekennzeichnet durch überbewegliche Gelenke, überdehnbare Haut und verletzliches Gewebe. Ursprünglich in sechs Haupttypen unterteilt, schlägt die revidierte Klassifikation von 2017 aufgrund neuer genetischer Erkenntnisse 13 Subtypen vor. Diese EDS-Subtypen basieren auf verschiedenen Defekten, insbesondere in Bezug auf Kollagen-Biosynthese, -Faltung und andere zelluläre Prozesse.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Wissenschaftlicher Hintergrund
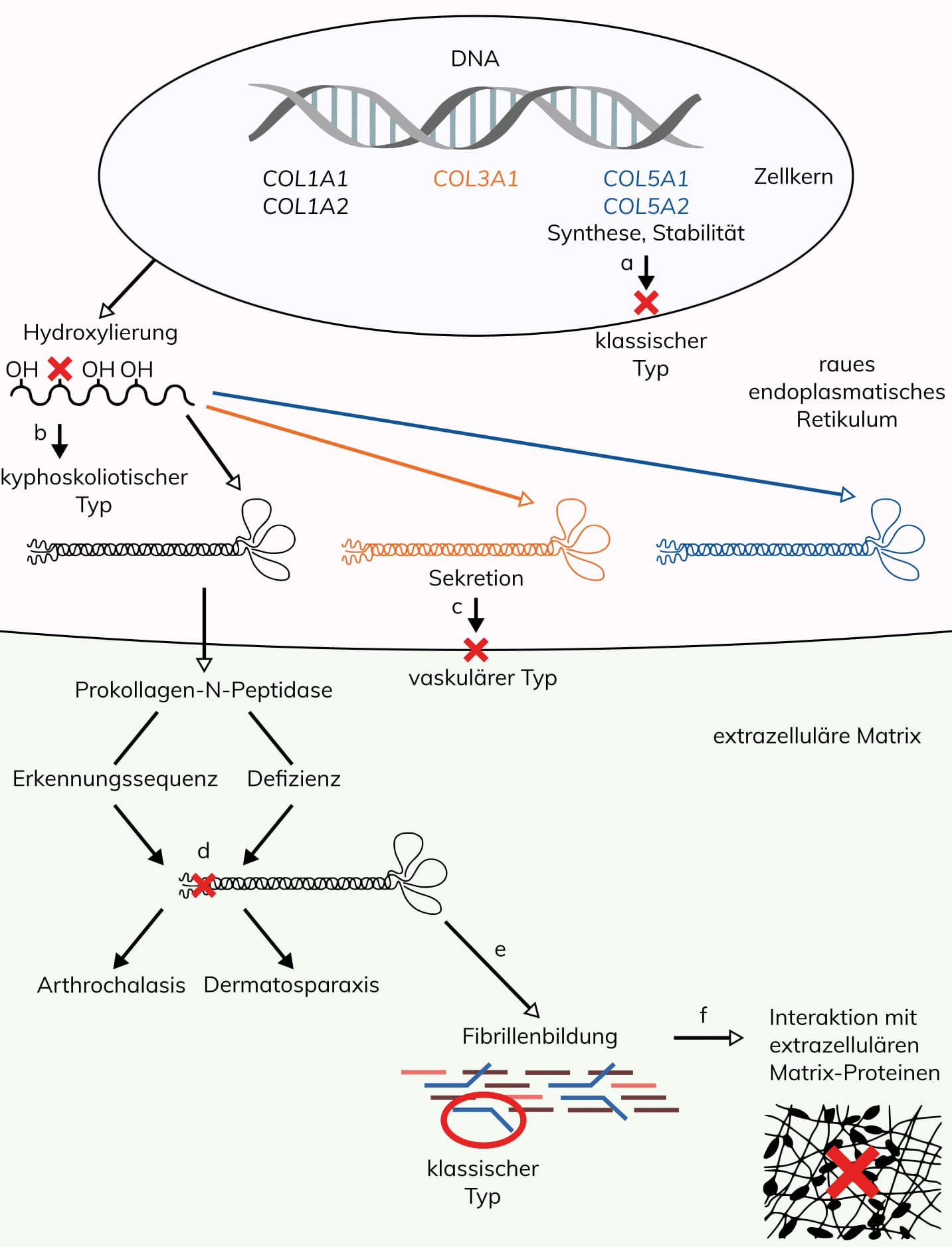
Unter Ehlers-Danlos-Syndrom (EDS) wird eine klinisch und genetisch heterogene Gruppe von Erkrankungen des Bindegewebes zusammengefasst, die durch eine Überbeweglichkeit der Gelenke, überdehnbare Haut und verletzliches Gewebe charakterisiert ist.
Nach der vereinfachten Villefranche-Klassifikation von 1998 wurde EDS anhand klinischer, biochemischer und genetischer Daten in sechs Haupttypen unterteilt, die auf verschiedene molekulare Defekte des Kollagenstoffwechsels zurückzuführen sind. Infolge der Zunahme von genetisch aufgeklärten, neuen EDS-Subtypen schlägt die revidierte internationale Klassifikation von 2017 anhand klinischer Kriterien 13 Subtypen vor, die autosomal-dominant oder rezessiv vererbt werden und mit Ausnahme des EDS hypermobiler Typ durch genetische Daten bestätigt werden können. Anhand genetischer Ursachen und pathogenetischer Mechanismen können die einzelnen EDS-Subtypen auf der Grundlage von Defekten der Kollagen-Biosynthese und -Prozessierung, der Kollagen-Faltung und Prozessierung, Defekten der Struktur und Funktion der Verbindung zwischen Muskel und der extrazellulären Matrix, Defekten der Glycosaminoglycan-Biosynthese, des Komplement-Systems und intrazellulärer Prozesse in verschiedene Gruppen unterteilt werden.