Alpha-Thalassämie
Die Alpha-Thalassämie ist eine genetisch bedingte Synthesestörung der α-Globinkette des Hämoglobinmoleküls. Die Erkrankung wird durch eine verminderte Produktion von Alpha-Globin-Ketten verursacht, was zu einem Überschuss an Beta-Globin-Ketten führt. Sie ist durch eine variabel ausgeprägte mikrozytäre, hypochrome Anämie ohne HbA2-Erhöhung gekennzeichnet. Ursächlich sind zumeist große Deletionen im Alpha-Globin-Genkomplex oder in selteneren Fällen pathogene Varianten im HBA1– oder HBA2-Gen.
Wissenschaftlicher Hintergrund
Die Alpha-Thalassämie beruht auf einer quantitativen Störung der α-Globinketten-Synthese. Durch das Defizit an α-Globinketten kommt es zur Bildung von Überschusshämoglobinen, die maßgeblich am Krankheitsbild der Alpha-Thalassämie beteiligt sind. Wie andere Hämoglobinopathien auch, tritt die Erkrankung gehäuft in Ländern auf, in denen Malaria endemisch ist und wird deshalb mit einer gewissen Resistenz gegenüber Plasmodien in Verbindung gebracht. Besonders häufig findet man die Alpha-Thalassämie in Völkern Asiens, Arabiens und Afrikas sowie den Mittelmeerländern (siehe Karte zur Prävalenz der Alpha-Thalassämie).
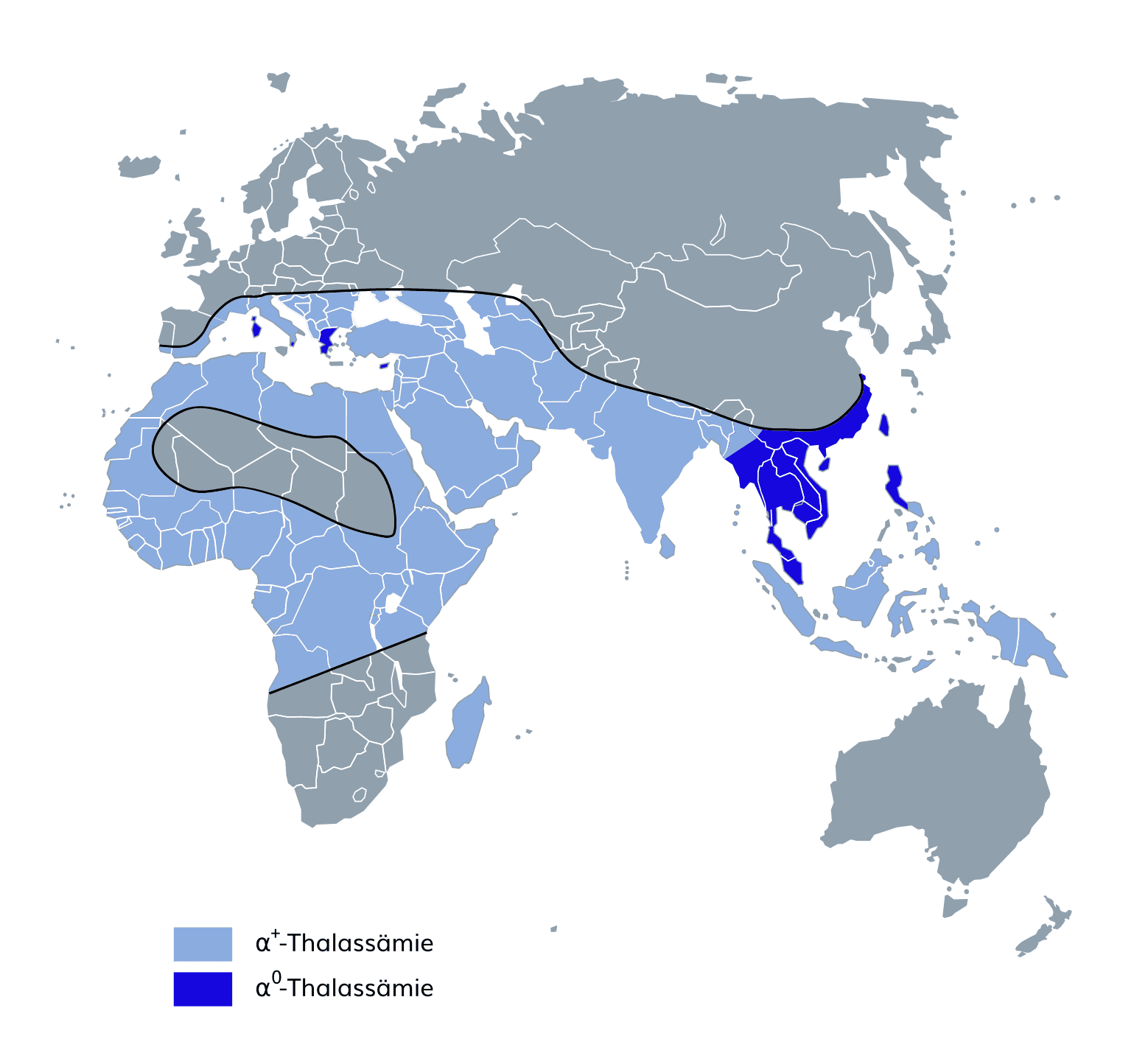
Die häufigste molekulargenetische Ursache für eine Alpha-Thalassämie ist die Deletion eines oder mehrerer α-Globin-Gene. Da in der normalen menschlichen Zelle der α-Globin-Genkomplex aus 4 α-Globin-Genen besteht (je ein HBA1– und ein HBA2-Gen auf jedem Chromosomen 16), ist der Schweregrad des Krankheitsbildes abhängig von der Anzahl der deletierten α-Globin-Gene. In seltenen Fällen können auch Punktmutationen bzw. kleinere Deletionen und Insertionen in den α-Globin-Genen für eine Alpha-Thalassämie ursächlich sein.
α-Thalassämie | Genotyp | Phänotyp | Hämatologie | Hb-Differenzierung | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Minima | -α/αα | unauffällig | Hypochromie und / oder Mikrozytose, selten mit Anämie | unauffällig | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Minor | -α/-α --/αα | meist unauffällig | Hypochromie, Mikrozytose, häufig mit Anämie | unauffällig | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HbH-Krankheit | --/-α | sehr variabel | hypochrome, hämolytische Anämie | HbH bis zu 40% (altersabhängig) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die meisten Träger einer Alpha-Thalassämie werden zufällig bei einer Routineuntersuchung entdeckt. Im fällt eine Mikrozytose und/oder eine Hypochromie – teilweise auch ein erniedrigter Hb-Wert – auf. Im Rahmen der Diagnostik sollte zuerst ein Eisenmangel durch die Bestimmung des ausgeschlossen werden, der die häufigste Ursache einer mikrozytären hypochromen Anämie darstellt. Im Vorfeld kann bereits durch die Huber-Herklotz-Formel abgeschätzt werden, ob es sich eher um eine Eisenmangelanämie oder eine Alpha-Thalassämie handelt.
HH = ((MCH * RDW) / (10 * Ery)) + RDW
Ein HH-Wert von < 20 spricht für alpha-Thalassämie. Ein HH-Wert von > 23 spricht für Eisenmangel.
Huber AR et al. Thalassämie-Syndrome: Klinik und Diagnose. Schweiz Med Forum 2004; 4: 947-52
Ist ein Eisenmangel ausgeschlossen, kann durch eine das Vorliegen einer untersucht werden. Häufig stellt sich die Frage nach der Anlageträgerschaft einer Hämoglobinopathie erst im Rahmen eines Kinderwunsches, entweder aufgrund einer positiven Familienanamnese oder durch die ethnische Herkunft. Bei einer bereits bestehenden Schwangerschaft ist eine zügige Diagnostik wichtig, um so früh wie möglich das Risiko einer schweren Form einer Hämoglobinopathie bei Nachkommen abschätzen zu können. Die detaillierte diagnostische Vorgehensweise zur allgemeinen Diagnostik von Hämoglobinopathien und zur Vorgehensweise bei Paaren mit Kinderwunsch ist in den folgenden interaktiven Flussdiagrammen dargestellt.
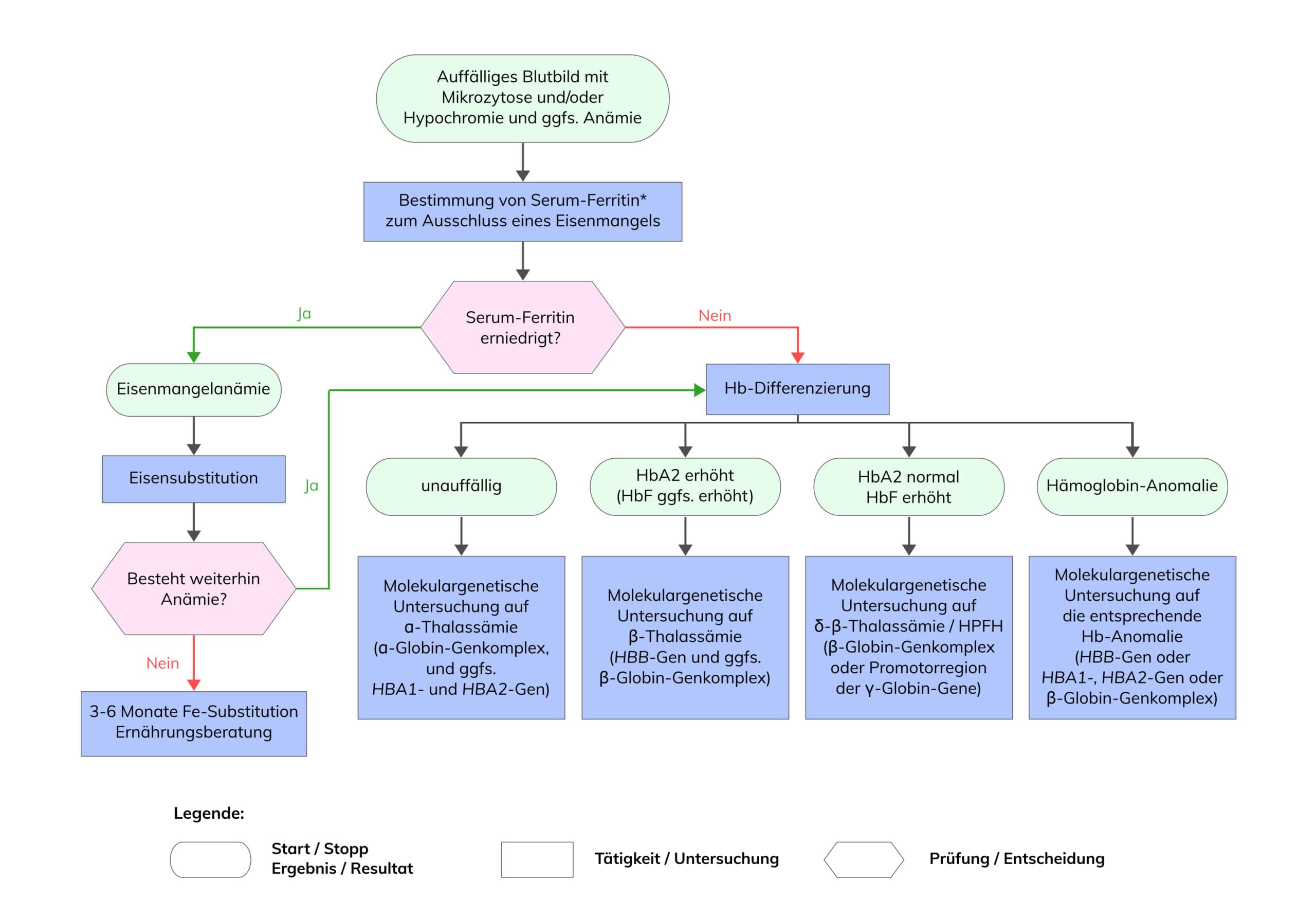
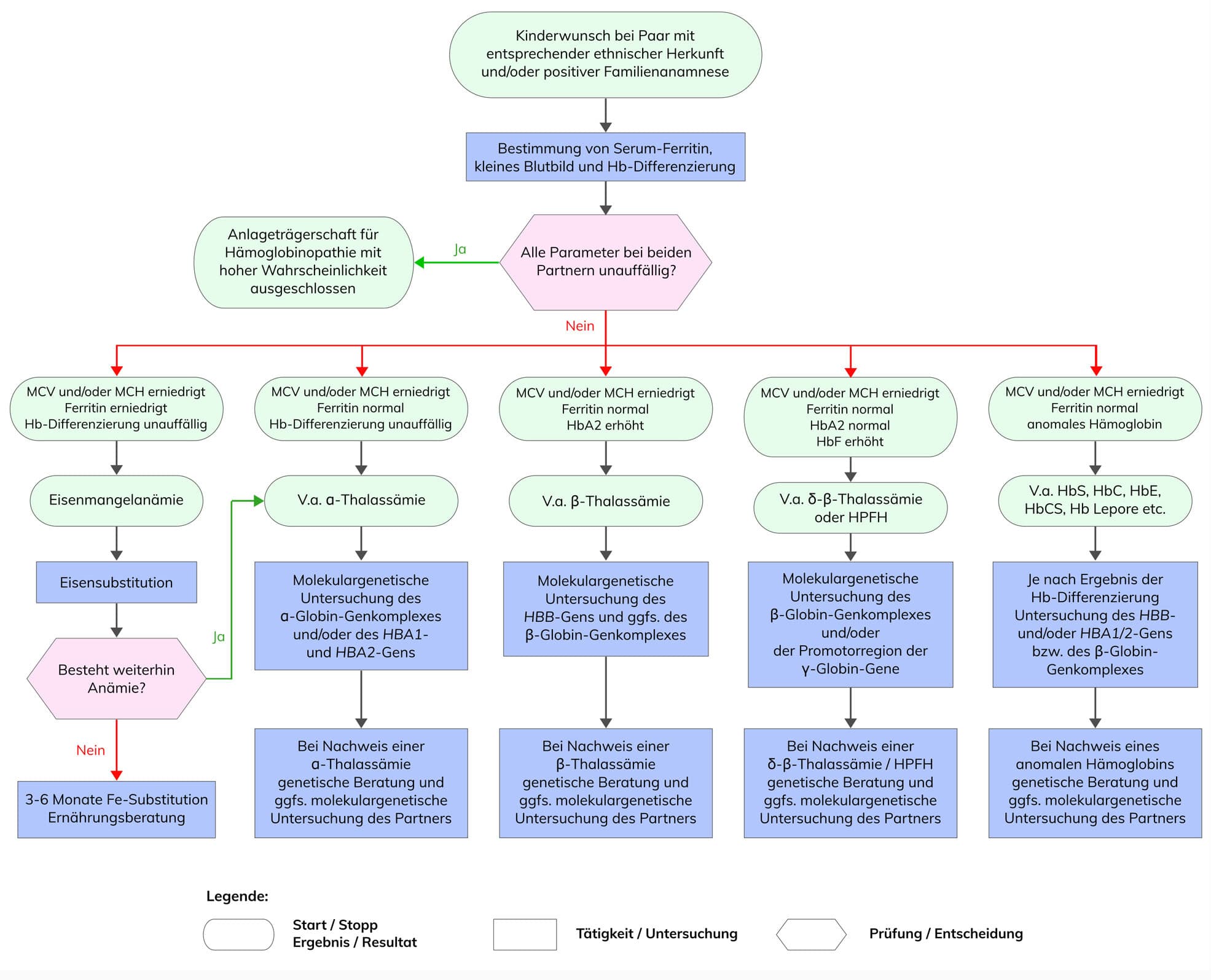
Da Hämoglobinopathien in den verschiedenste Kombinationen auftreten können, sollten die hämatologischen Befunde immer mit den Ergebnissen aus der Molekulargenetik abgeglichen und auf Validität geprüft werden.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.