Methoden & Technologien
DNA-Sequenzanalyse nach Sanger

Sanger-Sequenzierung
Die Sanger-Sequenzierung, benannt nach ihrem Entwickler Frederick Sanger, ist eine Methode zur Bestimmung der genauen Sequenzabfolge von Nukleotid-Basen in einem DNA-Strang. Seit ihrer Erfindung in den 1970er Jahren war sie bis zur Einführung des Next Generation Sequencing (NGS) der Goldstandard zum Nachweis von DNA-Varianten. Zur Durchführung wird neben einer DNA-Polymerase, Puffer und Primern, ein Nukleotid-Mix der vier in der DNA enthaltenen Basen sowie fluoreszenzmarkierte Stoppnukleotide (Dideoxy-Nukleotide) benötigt. Der Einbau der Stopp-Nukleotide führt in der Sequenzierreaktion zu einem Abbruch der DNA-Kettenbildung, so dass fluoreszenzmarkierte Produkte unterschiedlicher Länge entstehen, die in einem Sequenziergerät mittels Elektrophorese in einem Polyacrylamid-Gel aufgetrennt werden. Anhand der entsprechenden Fluoreszenzfarbe des ddNukleotids kann durch Laserlichtanregung unterschieden werden, welche Base am Ende der jeweiligen Kette eingebaut wurde. Damit lässt sich die Basenabfolge der sequenzierten DNA-Fragmente in einer entsprechenden Auswertesoftware auslesen und Abweichungen von der Referenzsequenz nachweisen.
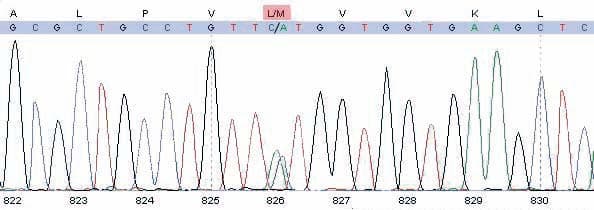
Heutzutage kommt die Sangersequenzierung vorwiegend dort zum Einsatz, wo NGS an technische oder bioinformatische Grenzen stößt und einen Genotyp nicht sicher auflösen kann (z.B. Pseudogenregionen, Repeat Expansionen, Homopolymere, komplexe Rearrangements , Hybridgene) oder wenn im Rahmen einer Zieldiagnostik einzelne bereits bekannte Varianten untersucht werden sollen (z.B. Segregationanalysen, Familienuntersuchung, Pränataldiagnostik). Die Vorteile liegen hier in der schnellen Bearbeitungszeit, da man die Proben problemlos einzeln ansetzen kann und nicht für einen NGS-Lauf sammeln muss. Zudem produzieren Sanger-Daten wenig Datenvolumen, da gerade bei Zelldiagnostik kein vollständiger NGS-Datensatz entstehen muss, um eine einzelne Variante auszuwerten.