Primäre Myelofibrose (PMF)
Die primäre Myelofibrose (PMF) ist eine Form der myeloproliferativen Neoplasie, die durch die Fibrose des Knochenmarkraumes und die Verdrängung der hämatopoetischen Aktivität gekennzeichnet ist. Sie verläuft in zwei Phasen: eine präfibrotische Phase mit hyperzellulärem Knochenmark und eine fibrotische Phase mit starker Fibrose und Osteosklerose. Genetische Varianten, insbesondere in den Genen JAK2, MPL und CALR, spielen eine entscheidende Rolle in der Pathogenese der PMF. Die Diagnose stützt sich auf spezifische WHO-Kriterien und der Krankheitsverlauf wird durch verschiedene Prognosemodelle, einschließlich MIPSS70 und GIPSS, bewertet.
Wissenschaftlicher Hintergrund
Die PMF gehört zu den myeloproliferativen Neoplasien und ist eine hämatopoetische Stammzellerkrankung mit Fibrose des Knochenmarkraumes und sukzessiver Verdrängung des hämatopoetisch aktiven Knochenmarks. Vorherrschend ist eine Proliferation der Megakaryozyten und der Granulozyten im Knochenmark. Sie entwickelt sich dabei allmählich aus einer frühen präfibrotischen Phase (präPMF), charakterisiert durch hyperzelluläres Knochenmark ohne oder mit minimaler Retikulinfibrose, in eine fibrotische Phase mit ausgeprägter Retikulin- oder Kollagenfibrose und häufig Osteosklerose.
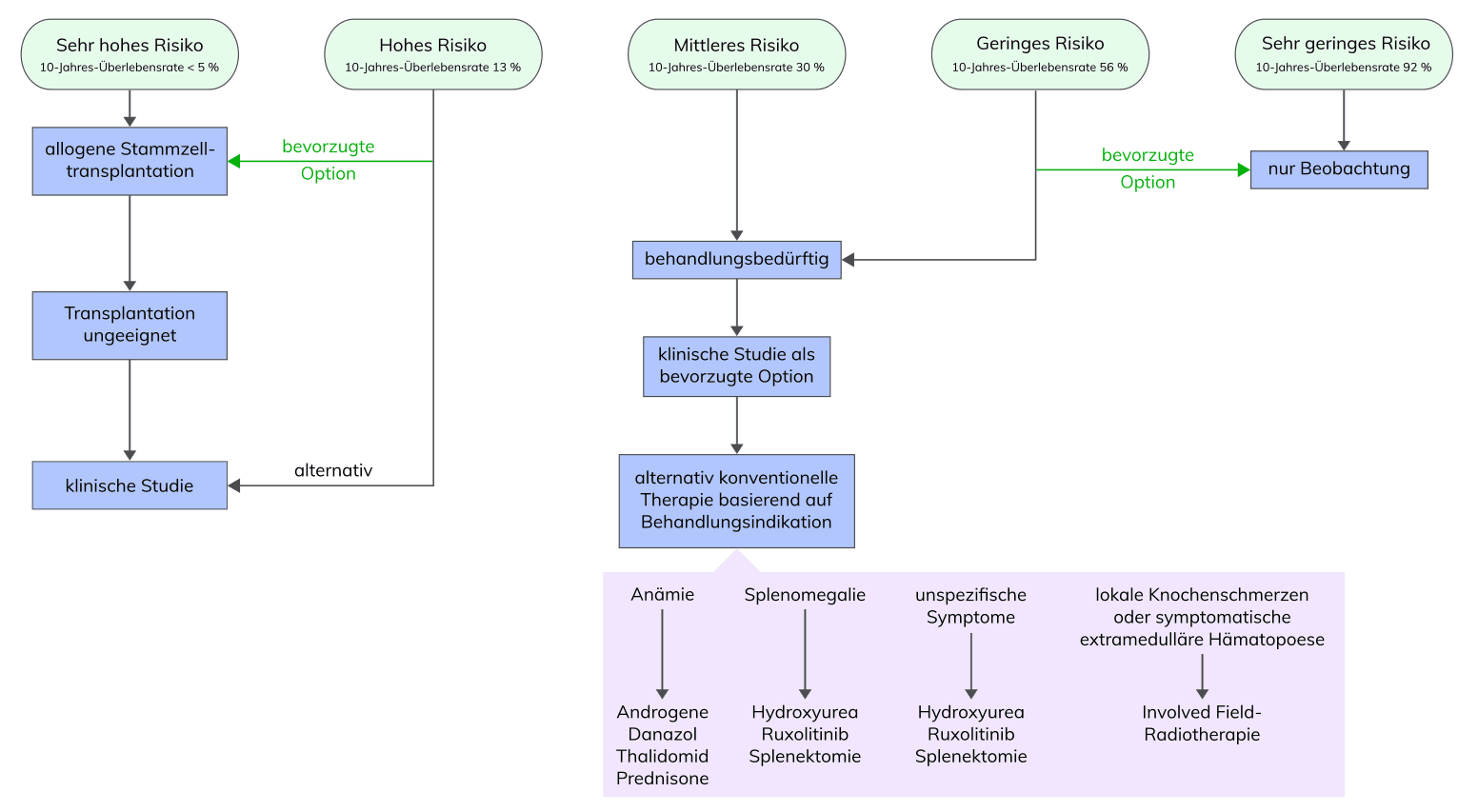
Das fortgeschrittene Stadium der PMF ist klinisch gekennzeichnet durch eine deutliche Anämie, grenzwertige bis milde Leukozytose oder Leukopenie, leichte Thrombozytose oder Verringerung der Thrombozytenzahl, hohe Serum-LDH-Level, das Vorhandensein von peripheren Erythro- und Myeloblasten (Leukoerythroblastose), tropfenförmigen Erythrozyten und Splenomegalie. Für den klinischen Verlauf ist eine Unterscheidung von und PräPMF wichtig. Eine Post-MF entwickelt sich bei 25-50% der Patienten mit und bei 2-3% der Patienten mit ET. Bei V.a. PMF sollte zunächst eine PV, ET, oder andere myeloproliferative Neoplasie ausgeschlossen werden.
Für die Diagnose einer PMF, präfibrotische Phase oder fibrotische Phase müssen alle drei Hauptkriterien und mindestens ein Nebenkriterium der entsprechenden WHO-Kriterien erfüllt sein.
PMF präfibrotische Phase | PMF fibrotische Phase |
| Hauptkriterien: 1. Megakaryozytäre Proliferation und Atypie, ohne Retikulinfibrose >Grad 1; begleitet von alters-angepasster Knochenmarkhyperzellularität, granulozytärer Proliferation und oftmals reduzierter Erythropoese 2. Kein Hinweis gemäß WHO auf BCR-ABL1+ CML, PV, ET, MDS oder andere myeloische Neoplasie 3. Nachweis einer JAK2, CALR oder MPL Variante oder bei fehlendem Nachweis dieser Varianten: Nachweis eines anderen klonalen Markers* oder Abwesenheit einer geringen reaktiven retikulären Knochenmarkfibrose** | Hauptkriterien: 1. Megakaryozytäre Proliferation und Atypie, begleitet von retikulärer und/oder kollagener Fibrose Grad 2 oder 3 2. Kein Hinweis gemäß WHO auf BCR-ABL1+ CML, PV, ET, MDS oder andere myeloische Neoplasie 3. Nachweis einer JAK2, CALR oder MPL Variante oder bei fehlendem Nachweis dieser Varianten: Nachweis eines anderen klonalen Markers* oder Abwesenheit einer reaktiven Myelofibrose** |
| Nebenkriterien: a. Anämie b. Leukozytose >11x109/L c. Splenomegalie d. Erhöhte Spiegel von Laktatdehydrogenase (LDH) | Nebenkriterien: a. Anämie b. Leukozytose >11x109/L c. Splenomegalie d. Erhöhte Spiegel von Laktatdehydrogenase (LDH) e. Leukoerythroblastose |
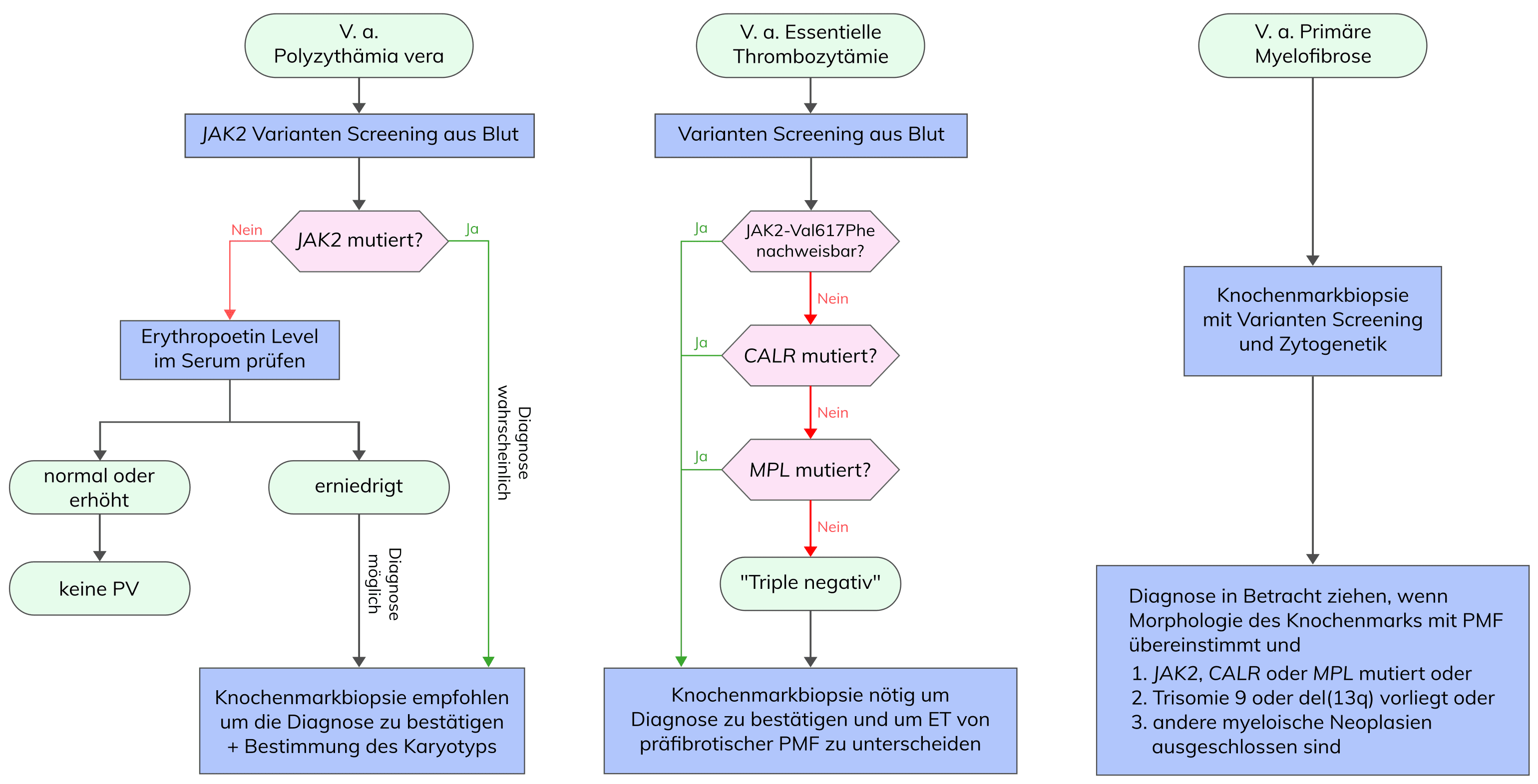
Somatische Varianten bei MPN, inklusive PMF, lassen sich in „Treiber“varianten und „andere“ Varianten klassifizieren. Zu den „Treiber“varianten zählen Varianten in JAK2, MPL und CALR, die sich in 90% der PMF-Patienten detektieren lassen. Die Variante JAK2 Val617Phe in Exon 14 wird in etwa 50-60% der PMF-Patienten nachgewiesen und führt zu einer konstitutiven Aktivierung des JAK-STAT-Signalweges.
In ca. 8% der PMF-Patienten können Varianten in Exon 10 von MPL, die hauptsächlich die Aminosäure Trp515 betreffen, nachgewiesen werden. Die häufigsten Varianten an MPL-Trp515 führen zu einem Aminosäureaustausch von Tryptophan nach Leucin (Trp515Leu), Lysin (Trp515Lys) oder Alanin (Trp515Ala) und somit ebenfalls zu einer konstitutiven Aktivierung des JAK-STAT-Signalweges.
Weitere ca. 30% der PMF-Patienten tragen Varianten in Exon 9 von CALR, die mit einem jüngeren Alter, höheren Thrombozytenwerten und niedrigerer Frequenz an Anämie, Leukozytose und Spliceosom-Varianten assoziiert sind. Bei den CALR-Varianten handelt es sich um verschiedene somatische Deletions- bzw. Insertionsvarianten, die zu einem Frameshift und somit zu einem spezifischen, alternativen Leserahmen führen. Man unterscheidet dabei vorrangig Typ 1- (52 bp-Deletionen, p.(L367fs*46)) bzw. Typ 1-ähnliche Varianten und Typ 2- (5 bp-Insertion, p.(K385fs*47)) bzw. Typ 2-ähnliche Varianten in CALR, wobei in PMF Typ 2- bzw. Typ 2-ähnliche Varianten mit einer erhöhten Risikogruppe, zirkulierendem Blastenzellanteil, Leukozytenzahl und vermindertem Überleben assoziiert sind. Bemerkenswert ist der Überlebensvorteil von PMF-Patienten mit CALR Typ 1- bzw. Typ 1-ähnliche Varianten gegenüber PMF-Patienten mit anderen Genotypen. CALR codiert für ein Ca2+-bindendes Chaperon des endoplasmatischen Retikulums.
PMF-Patienten, die keine Variante in JAK2, MPL oder CALR aufweisen, werden als triple-negativ bezeichnet. In Einzelfällen kann bei diesen Patienten eine SH2B3-Variante nachgewiesen werden. SH2B3 spielt eine entscheidene Rolle in der Hämatopoese durch negative Regulation der JAK2-Aktivierung. Die häufigsten Varianten finden sich in Exon 2 des SH2B3-Gens, gefolgt von Exon 8.
Allgemein konnte gezeigt werden, dass PMF-Patienten mit Varianten in einem dieser Gene – ASXL1, EZH2, SRSF2, IDH1, IDH2, U2AF1 – eine IPSS/DIPSS-plus unabhängige molekulare Hochrisikogruppe mit einem kürzeren Gesamtüberleben und höherem AML-Risiko darstellen als PMF-Patienten ohne Varianten in diesen Genen. Dabei zeigen CALR+/ASXL1- Patienten das längste Überleben, während CALR-/ASXL1+ Patienten das kürzeste Überleben zeigten.
Varianten in RAS und CBL sind mit einem schlechten Ansprechen auf eine Therapie mit Ruxolitinib, schlechten prognostischen Eigenschaften sowie einem vermindertem Überleben assoziiert.
Zytogenetische Veränderungen sind bei ca. 40% der Patienten nachweisbar. Das Vorliegen einer del(13)(q12-22), einer der(6)t(1;6)(q21-23;p21.3) oder einer Trisomie 9 ist dabei stark mit einer PMF assoziiert, aber nicht pathognomonisch für eine PMF.
Es lassen sich drei zytogenetische Risikogruppen unterscheiden:
- günstig: normaler Karyotyp, Einzelaberrationen von 20q-, 13q-, +9, Chromosom 1 Translokation/Duplikation und Veränderungen der Geschlechtschromosomen inkl. –Y
- sehr ungünstig (very high risk (VHR)): Einzel- oder Mehrfachaberrationen von -7, inv(3)/3q21, i(17q), 12p-/12p11.2, 11q-/11q23, Trisomien der Autosomen (ohne +8, +9)
- ungünstig: alle anderen Veränderungen.
Auf Basis dieser Veränderungen wurden drei neue Prognosemodelle bei der PMF vorgeschlagen: