Polyzythämia vera (PV)
Die Polyzythämia vera (PV) ist eine myeloproliferative Neoplasie, die durch eine erhöhte Erythropoese und eine klonale Expansion hämatopoetischer Stammzellen charakterisiert ist. Das Risiko für thromboembolische Komplikationen ist deutlich erhöht, besonders bei älteren Patienten und solchen mit vorherigen Thrombosen. Diagnostisch ist der Nachweis einer JAK2-Variante (Val617Phe oder Exon 12) entscheidend, die fast alle PV-Patienten tragen und eine unkontrollierte Blutzellproliferation verursacht. Die Krankheit verläuft in zwei Phasen: einer anfänglichen polyzythämischen Phase, gefolgt von einer post-polyzythämischen Myelofibrose-Phase, die zu Knochenmarkfibrose und Splenomegalie führt.
Wissenschaftlicher Hintergrund
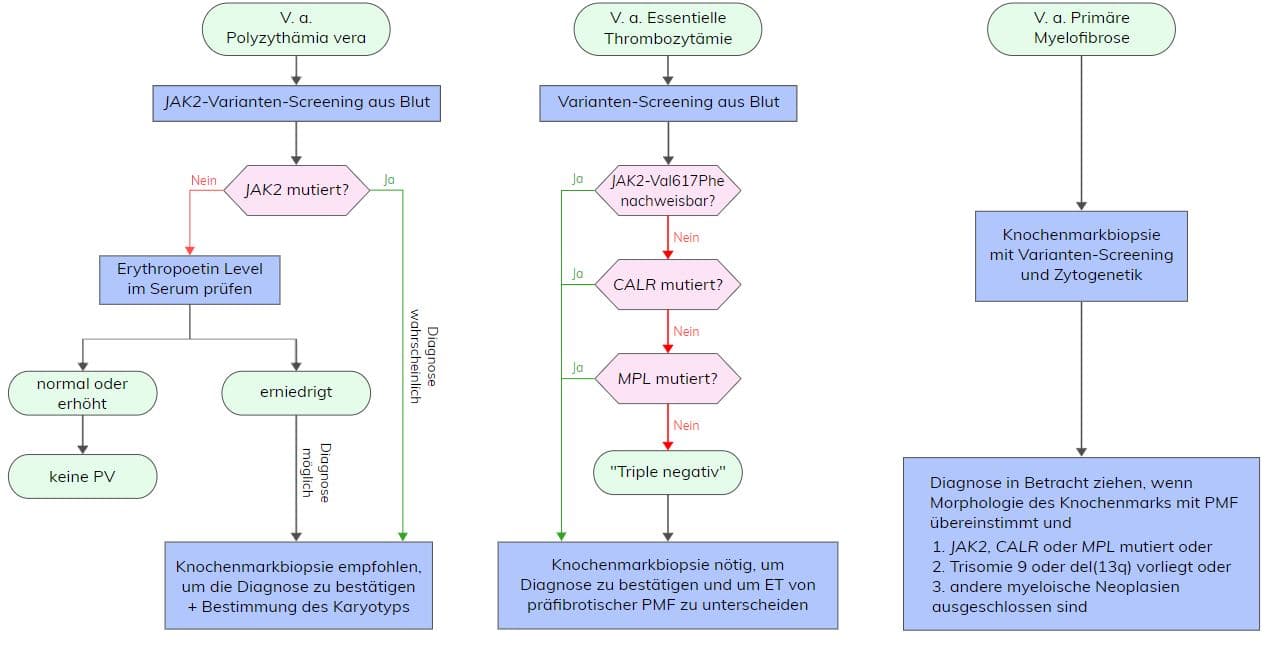
Die Polyzythämia vera (PV) gehört zu den myeloproliferativen Neoplasien (MPN) und ist eine hämatopoetische Stammzellerkrankung, charakterisiert durch eine klonale Expansion mit Betonung der Erythropoese. Die Inzidenz erhöht sich mit zunehmendem Alter und variiert von 0,01-2,8 Fälle/100.000/Jahr in Europa und Nordamerika. Der Krankheitsverlauf ist durch das deutlich gesteigerte Risko für potenziell lebensbedrohliche arterielle und venöse thromboembolische Komplikationen gekennzeichnet. Hauptrisikofaktoren für Gefäßkomplikationen sind höheres Lebensalter und eine bereits stattgehabte Thrombose. Verwandte ersten Grades haben ein fünf- bis siebenfach erhöhtes Risiko für die Entwicklung einer MPN. Klinisch ist die PV durch zwei Phasen bestimmt: die polyzythämische Phase mit erhöhten Hämoglobin-Leveln und erhöhtem Hämatokrit sowie vermehrter Erythrozytenmasse, die bis zu 20 Jahre bestehen kann. Darauf folgt eine progrediente post-polyzythämische Myelofibrose-Phase(post-PV Myelofribrose) mit zunehmender Zytopenie, Knochenmarkfibrosen, extramedullärer Blutbildung und Splenomegalie. Etwa 10% der Patienten zeigen einen Übergang in ein MDS oder eine AML. Gerade in der frühen Phase der PV kann eine Abgrenzung zur ET oder PMF schwierig sein. Für die Diagnose einer PV müssen alle drei Hauptkriterien oder die ersten beiden Hauptkriterien und das Nebenkriterium der WHO erfüllt sein.
Hauptkriterien
- Hämoglobin > 16,5 g/dL bei Männern, > 16,0 g/dL bei Frauen oder Hämatokrit > 49% bei Männern, > 48% bei Frauen
- Trilineäre Hyperzellularität mit gesteigerter Erythropoese, Granulopoese und Megakaryopoese mit pleomorphen Megakaryozyten im Knochenmark
- Nachweis einer JAK2 Val617Phe-Variante oder einer JAK2 Exon 12-Variante
Nebenkriterien
- Niedriger Erythropoetin-Spiegel
Etwa 96% der Patienten mit PV tragen eine Variante in Exon 14 des JAK2-Gens (Val617Phe) und etwa 3% eine Variante in Exon 12 des JAK2-Gens, so dass nahezu alle Patienten mit PV eine JAK2-Variante aufweisen, die als Auslöser der unkontrollierten Myeloproliferation gelten. Das entstehende Protein verfügt über eine gesteigerte Tyrosinkinase-Aktivität. Die Janus-Kinase 2 ist bei Gesunden essentiell für die Blutbildung. Über sie führen Zytokine wie Erythropoetin, Thrombopoetin oder G-CSF zu einer Aktivierung der STAT-Moleküle in der Zelle und zu einer Proliferation hämatopoetischer Zellen. Eine Variante in JAK2 führt zu einer Liganden-unabhängigen, konstitutiven Signaltransduktion. Hierdurch kommt es zu einer unkontrollierten Proliferation von Blutzellen. Eine hohe JAK2 Val617Phe-Varianten-Allel-Frequenz (VAF) ist mit Pruritus und einer fibrotischen Transformation assoziiert. Generell zeigen Patienten mit dieser Variante meist ein höheres Alter, höhere Hämoglobinwerte, Leukozytose und geringere Thrombozytenwerte. Patienten mit einer JAK2 Exon 12-Variante haben eher eine prädominante erythroide Myelofibrose, niedrige Erythropoetin-Level und ein jüngeres Alter bei Diagnosestellung.
Nur bei wenigen PV-Patienten kann eine Variante in MPL oder CALR detektiert werden.
Etwa 53% der PV-Patienten zeigen eine oder mehr Varianten, unabhängig von JAK2/MPL/CALR, wobei Varianten in TET2 und ASXL1 am häufigsten sind. Ungünstige Varianten bzgl. des Gesamt-, Leukämie- oder Fibrose-freiem Überleben werden bei PV in den Genen ASXL1, SRSF2 und IDH2 detektiert.
Ein weiterer Marker ist die Überexpression des PRV1-Gens, die bei etwa 80% der PV-Patienten nachweisbar ist. Sie korreliert mit dem Auftreten von erhöhten Erythropoeitin-unabhängigen, erythroiden Kolonien (EEC) und ist hilfreich bei der Diskriminierung von PV und ET zu sekundärer Erythrozytose und Thrombozytose.
Bei ca. 20% der Patienten sind bei Diagnosestellung bereits zytogenetische Veränderungen nachweisbar (v.a. Trisomie 8 und 9, Deletionen im langen Arm von Chr. 20 und 13 bzw. im kurzen Arm von Chr. 9). Die Veränderungen sind progredient und werden in 80-90% der Fälle mit post-PV-Myelofibrose beobachtet.
Risikofaktoren für eine leukämische Transformation sind fortgeschrittenes Alter, Leukozytose und eine veränderter Karyotyp.