Morbus Fabry
Morbus Fabry ist eine X-chromosomal vererbte Störung des Glycosphingolipid-Katabolismus, die durch eine Defizienz der lysosomalen Alpha-Galaktosidase A bedingt ist. Ursächlich sind pathogene Varianten im GLA-Gen. Der Enzymdefekt führt zu einer fortschreitenden systemischen Akkumulation von Glycosphingolipiden in verschiedenen Geweben und Organen, die im weiteren Krankheitsverlauf zu Schlaganfall, Herzinfarkt und Dialysepflicht führen können.
Wissenschaftlicher Hintergrund
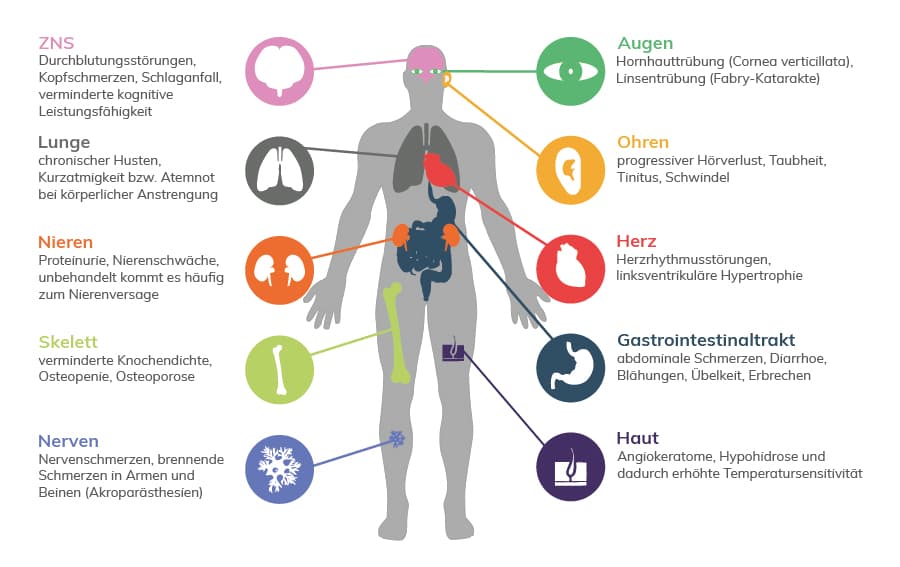
Morbus Fabry zählt zu den lysosomalenSpeicherkrankheiten und ist eine angeborene, X-chromosomal vererbte Störung des Glycosphingolipid-Katabolismus, die durch eine herabgesetzte bzw. fehlende Aktivität des lysosomalen Enzyms Alpha-GalaktosidaseA (GLA) bedingt ist. Ursächlich hierfür sind Varianten im GLA-Gen. Der Enzymdefekt führt zu einer fortschreitenden systemischen Akkumulation von Glycosphingolipiden in verschiedenen Geweben und Organen. Zur Vermeidung schwerwiegender Komplikationen ist eine frühzeitige Diagnosestellung zur Therapieeinleitung von großer Bedeutung. Zu den Symptomen der Fabry-Erkrankung zählen Angiokeratome, Schmerzattacken, Funktionsstörungen verschiedener Organe, die im weiteren Krankheitsverlauf zu Schlaganfall, Herzinfarkt und Dialysepflicht führen können. Zeitpunkt der Erstmanifestation und Verlauf der Erkrankung sind hochvariabel, häufig beginnen die Beschwerden bereits im Kindesalter.
Die Inzidenz des klassischen Morbus Fabry bei Männern wird auf ca. 1:40.000 geschätzt. Im Gegensatz zu den meisten anderen X-chromosomal vererbten Erkrankungen sind heterozygote Frauen selten asymptomatisch und können behandlungsbedürftige Symptome bis hin zum Vollbild der Erkrankung entwickeln. Eine Hypothese für die phänotypische Variabilität bei heterozygoten Frauen ist die verschobene X-Inaktivierung.
Seit 2001 besteht in Europa die Möglichkeit einer Enzymsubstitutionstherapie. Seit 2016 besteht alternativ für Patienten über 16 Jahren mit gesichertem Morbus Fabry die Möglichkeit einer oral einzusetzenden Chaperon-Therapie mit dem Wirkstoff . Da die Therapie nur bei Vorliegen bestimmter Varianten im GLA-Gen wirksam ist, muss der GLA-Genotyp des Patienten bekannt sein. Eine Liste der einzelnen Varianten findet sich in der Fachinformation Galafold.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Indikation | ICD—10 | Gen | OMIM—G |
| Morbus Fabry | E75.2 | GLA | 300644 |