MODY (Maturity-onset diabetes of the young)
Der MODY-Diabetes (Maturity-onset Diabetes of the Young) ist eine autosomal-dominant vererbte Diabetesform, die bis zu 5% aller Diabetesfälle in Europa ausmacht und meist vor dem 25. Lebensjahr auftritt. MODY wird oft fälschlicherweise als Typ 1 oder Typ 2 Diabetes diagnostiziert und kann durch spezifische diagnostische Kriterien, wie den negativen Antikörpernachweis und den Auftreten bestimmter Symptome, identifiziert werden. Es gibt derzeit 14 klassifizierte MODY-Typen, basierend auf ihrer Klinik und den betroffenen Genen, wobei Typ 2 (GCK) und 3 (HNF1A) am häufigsten vorkommen.
Wissenschaftlicher Hintergrund
„Maturity-onset Diabetes of the Young“ (MODY) bezeichnet eine autosomal-dominant vererbte Gruppe klinisch heterogener, nicht immer insulinabhängiger Formen des Diabetes, die durch verschiedene Störungen der Betazell-Funktionen im Pankreas charakterisiert ist.
MODY ist die häufigste Form des monogenen Diabetes und ist für bis zu 5% aller diabetischen Erkrankungen in Europa verantwortlich. Die Erkrankung wird meist vor dem 25. Lebensjahr entdeckt und oftmals jedoch zunächst als Diabetes mellitus Typ 1 oder Typ 2 diagnostiziert. Können bei Normalgewichtigen mit diabetischer Stoffwechsellage jedoch keine Antikörper gegen GAD, IA-2 und / oder Inselzellen nachgewiesen werden, sollte ein MODY in Betracht gezogen werden. Bei Auftreten eines Gestationsdiabetes sollte ebenfalls an einen MODY gedacht werden, der in ca. 25% der betroffenen Schwangerschaften nachgewiesen werden kann.
Diagnostische Kriterien für die Verdachtsdiagnose MODY sind:
- Manifestation im Jugendalter oder frühe Adoleszenz (< 35 Jahre)
- Antikörpernachweise GAD, IA-2 und/oder Inselzellen negativ
- Typ 1 und Typ 2 Diabetes oder metabolisches Syndrom ausgeschlossen
- moderate (Nüchtern-) Hyperglykämie (130-250 mg/dl, oder 7-14 mM) vor dem 30. Lebensjahr
- positiver Glukose-Belastungstest
- Schwangerschaftsdiabetes
- permanent niedriger Insulinbedarf (z.B. <0,5 U/kg/d)
- zystische Nierenerkrankungen beim Patienten (oder nahen Verwandten)
- Glukosurie
- betroffener Verwandter 1. Grades
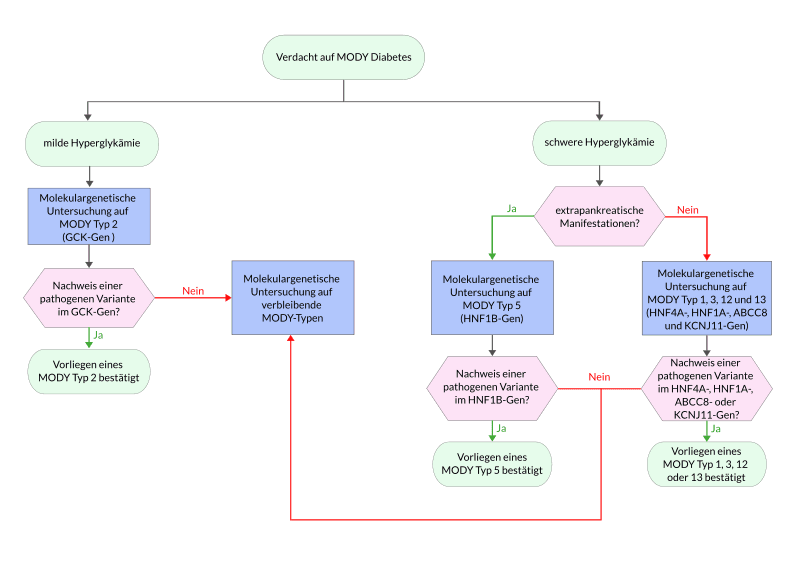
Die verschiedenen Formen des MODY werden nach ihrer Klinik und den entsprechenden von pathogenen Varianten betroffenen Genen klassifiziert. Derzeit werden 14 Typen unterschieden, wobei MODY Typ 2 und 3 die häufigsten Formen darstellen.
MODY Typ 1, 3, 12 und 13
MODY Typ 1, 3, 12 und 13 weisen einen klinisch ähnlichen Phänotyp auf und sind durch eine ausgeprägte progrediente Hyperglykämie gekennzeichnet. Die betroffenen Patienten sprechen auf eine Therapie mit niedrig dosierten Sulfonylharnstoffen sehr gut an, fallen jedoch unter Therapie durch überdurchschnittlich häufige Episoden von Hypoglykämie auf. Ursächlich sind Varianten in den Genen HNF4A, HNF1A, die für Transkriptionsfaktoren codieren sowie ABCC8 und KCNJ11, die die Untereinheiten des ATP-sensitiven Kaliumkanals bilden. Die molekulargenetische Untersuchung der Gene ABCC8 und KCNJ11 sollte in Betracht gezogen werden, wenn der Patient auf Sulfonylharnstoffe anspricht und MODY Typ 1 und Typ 3 bereits ausgeschlossen wurden.
MODY Typ 2
MODY Typ 2 weist eine persistierende, milde Hyperglykämie auf, die in der Regel keiner medikamentösen Therapie bedarf und durch eine entsprechende Diät gut zu behandeln ist. Die Erkrankung wird durch pathogene Varianten im Glukokinase-Gen (GCK) verursacht. Da der MODY Typ 2 mit einer sehr milden Symptomatik einhergeht, wird er in vielen Fällen nur zufällig im Rahmen einer Routinediagnostik entdeckt, wie z.B. bei Schwangeren während des Screenings auf eine gestörte Glucosetoleranz.
MODY Typ 4
MODY Typ 4 zählt zu den seltenen MODY-Formen und ist bedingt durch pathogene Varianten im Gen für den Transkriptionsfaktor PDX-1. Er ist aufgrund der nur leichten Hyperglykämie mit einem milden Krankheitsverlauf assoziiert und ähnelt phänotypisch dem MODY Typ 2.
MODY Typ 5
MODY Typ 5 kann neben der ausgeprägten Hyperglykämie zusätzlich eine oder auch Fehlbildungen des Urogenitaltrakts zeigen, wodurch eine deutliche Abgrenzung zu den übrigen MODY-Formen möglich ist. MODY Typ 5 wird durch pathogene Varianten im Gen für den Transkriptionsfaktor HNF-1B verursacht.
MODY Typ 6-11 und 14
Eine eindeutige Beschreibung zur klinischen Symptomatik bei den MODY Typen 6-11 und 14 (NEUROD1, KLF11, CEL, PAX4, INS, BLKund APPL1 ) ist aufgrund der Seltenheit (< 1%) bisher nicht möglich.
Da nicht bei allen Patienten mit einem MODY ursächliche Varianten in den entsprechenden Genen gefunden werden, geht man davon aus, dass es noch weitere bisher unbekannte mit MODY assoziierte Gene gibt.
Übersicht über die verschiedenen Formen des MODY-Diabetes:
MODY Typ | Gen | Funktion des Genprodukts | Symptome / weitere Manifestationen | Therapie | Häufigkeit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MODY 1 | HNF4A | Transkriptionsfaktor, reguliert HNF1A-/PDX1-Transkription und weitere Gentranskripte | deutlich progressive Hyper- glykämie; niedrige Trigly- ceride und Apolipoproteine | Diät, Sulfonylharnstoffe, Insulin | 5-10% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MODY 2 | GCK | Glukokinase, katalysiert Reaktion Glukose → Glukose-6-Phosphat | milde Hyperglykämie; "Gestationsdiabetes" | Diät, Bewegung, (ggfs. Insulin in Schwangerschaft) | 30-50% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MODY 3 | HNF1A | Transkriptionsfaktor, reguliert u.a. Insulin-Gentranskription | deutlich progressive Hyperglykämie; progressiver Insulinsekretions- defekt; Glukosurie | Diät Sulfonylharnstoffe, Insulin | 30-65% | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.