Long QT-Syndrom (LQTS)
Das Long QT-Syndrom (LQTS) ist eine genetische Herzerkrankung, die durch eine verlängerte ventrikuläre Repolarisation und eine erhöhte Anfälligkeit für lebensbedrohliche Arrhythmien gekennzeichnet ist. Bei bis zu 95% der diagnostizierten Fälle sind pathogene Varianten in den Genen KCNQ1, KCNH2 und SCN5A beteiligt. Die Erkrankung kann verschiedene Formen aufweisen und die frühzeitige Identifizierung von Anlageträgern ermöglicht präventive Behandlungsstrategien, einschließlich der Vermeidung bestimmter Medikamente und der Einnahme von β-Blockern.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Wissenschaftlicher Hintergrund
Das Long QT-Syndrom (LQTS) ist eine klinisch und genetisch heterogene Herzerkrankung, die durch eine verlängerte ventrikuläre Repolarisation charakterisiert ist. Im Langzeit-EKG lässt sich eine verlängerte Frequenz-korrigierte QT-Zeit (QTc) von 460 bis >500 ms nachweisen. Auf molekularer Ebene ist das verlängerte QTc-Intervall die Folge eines Rückgangs der K+-Auswärtsströme (hauptsächlich IKs, IKr und IK1) oder der Anstieg der Einwärtsströme (hauptsächlich INa und ICaL). In Abhängigkeit von der QTc kommt es zu Arrhythmien, die zu Bewusstlosigkeit und plötzlichem Herztod führen können. Die 10-Jahres-Mortalität beträgt unbehandelt 50%. Man unterscheidet die häufige autosomal-dominanteRomano-Ward-(RW) und die sehr seltene autosomal-rezessiveJervell-Lange-Nielsen-Form(JLN). Die Prävalenz des Long QT-Syndroms in der kaukasischen Bevölkerung ist mindestens 1:2.500.
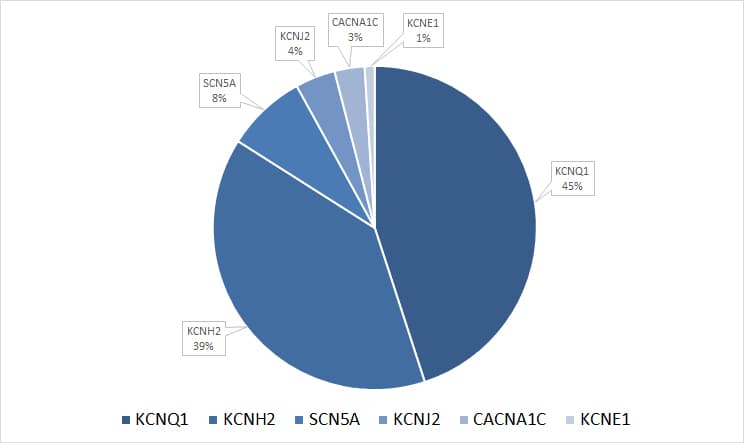
Als cutoff für die genetische Diagnostik sollte postpubertär eine QTc >470 ms für Männer, >480 ms für Frauen sowie >460 ms für Kinder dienen, da dieses Kollektiv von der Diagnostik profitieren kann. In ca. 90-95 % der molekulargenetisch positiven LQTS-Fälle werden pathogene Varianten in einem der drei Gene KCNQ1, KCNH2 und SCN5A nachgewiesen. KCNQ1 und KCNH2 codieren für die kardialen Kaliumkanäle Kv7.1 und Kv11.1. Während Kv7.1 den IKs-Strom (langsam verzögert gleichrichtender K+-Strom) vermittelt, ist Kv11.1 für den IKr-Strom (schnell verzögert gleichrichtender K+-Strom) verantwortlich. Beide Kanäle tragen zur Repolarisation der Zelle bei und beenden somit das kardiale Aktionspotential. Das SCN5A-Gen codiert für den Nav1.5-Kanal, welcher den Einstrom von Natriumionen (INa) über die Zellmembran vermittelt und somit an der schnellen Depolarisierung des kardialen Aktionspotentials beteiligt ist. Ein pathologisch reduzierter Kaliumstromfluss bzw. ein pathologisch erhöhter Natriumstromfluss kann die Dauer der ventrikulären Repolarisation und damit die QTc verlängern. In etwa 25% der klinisch gesicherten LQTS-Fälle kann keine genetische Ursache nachgewiesen werden. Die molekulare Klassifikation und Nomenklatur orientiert sich hierbei an den betroffenen Genen:
- KCNQ1 (LQTS Typ1; 45 % der pathogenen Varianten, eigene Daten) Varianten mit dominanter oder rezessiver Ausprägung (RW- und JLN-Form) sind beschrieben. Patienten mit pathogenen Varianten im KCNQ1-Gen zeigen meist frühzeitig einen deutlich ausgeprägten Phänotyp mit einem hohen Risiko für kardiale Ereignisse.

- KCNH2 (LQTS Typ2; 39 % der pathogenen Varianten, eigene Daten) Es besteht ein hohes Risiko für kardiale Ereignisse. Es handelt es sich um RW-Formen.
- SCN5A (LQTS Typ3; 8 % der pathogenen Varianten, eigene Daten) Kardiale Ereignisse sind seltener, die Letalität ist jedoch fünffach höher. Klinisch tritt LQTS Typ 3 als RW-Form auf.
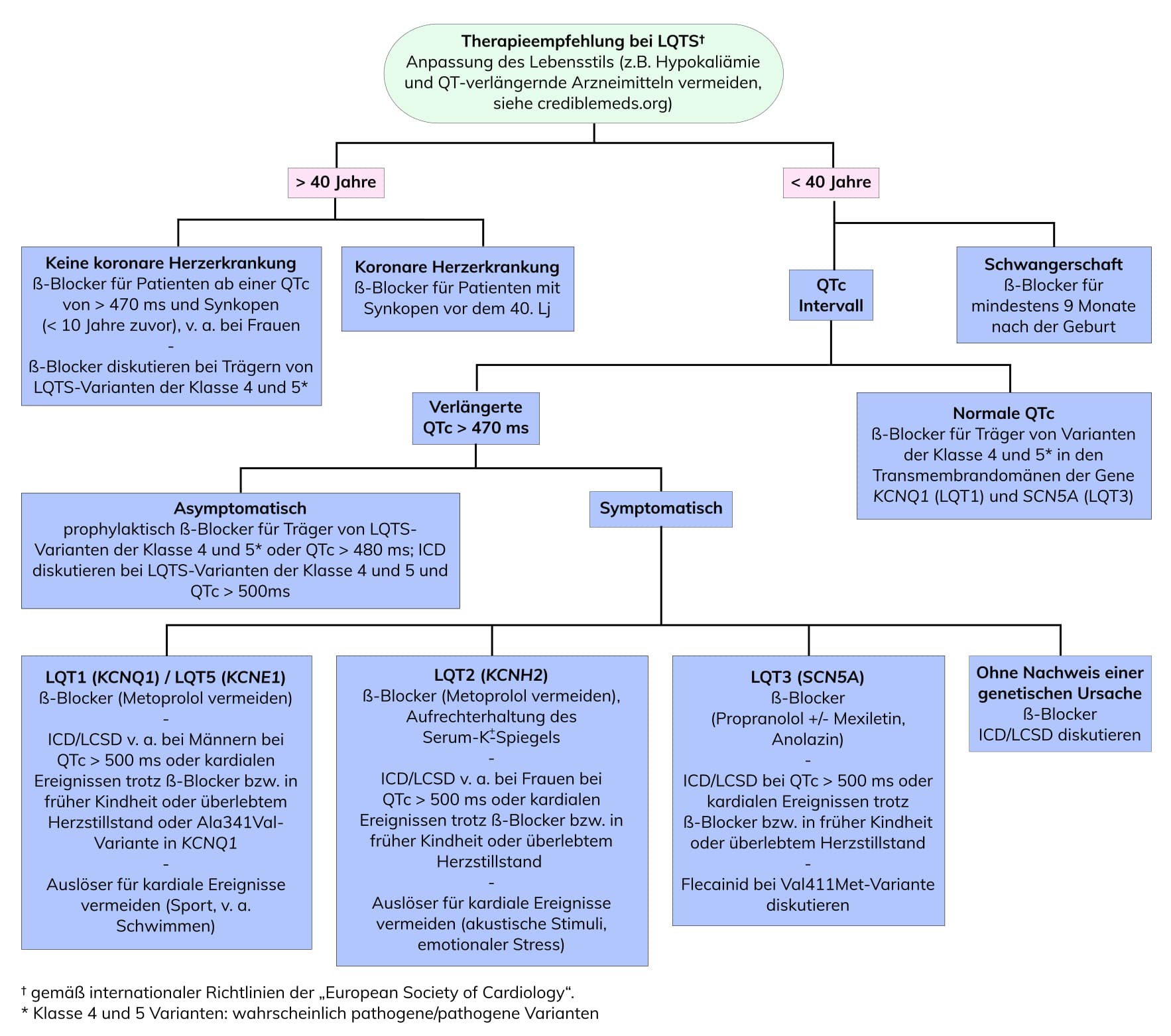
Die Identifikation von Anlageträgern pathogener Varianten ermöglicht eine rechtzeitige, ggfs. präsymptomatische Therapie. Das Risiko für kardiale Ereignisse wird dadurch beim LQTS Typ 1 um 62-95% und beim LQTS Typ 2 um 74% reduziert. Gemäß internationaler Leitlinien sollten alle LQTS-Patienten ab dem Zeitpunkt der Diagnose den Lebensstil anpassen (z.B. die Vermeidung von QT-verlängernden Arzneimitteln, siehe ) und mit β-Blockern behandelt werden. Die Implantation eines ICD wird bei therapierefraktären rezidivierenden Synkopen / ventrikulären Tachykardien, einem vorangegangenen Herzstillstand oder bei LQT2-Patienten mit einer QTc von >500 ms empfohlen.
Darüber hinaus wurden bei seltenen Sonderformen des Long QT-Syndroms, die durch spezielle z.T. komplexe Phänotypen gekennzeichnet sind, pathogene Varianten in weiteren Genen identifiziert, deren Analyse in einzelnen Familien sinnvoll sein kann:
- KCNE1 (LQTS Typ 5 / Jervell und Lange-Nielsen Syndrom 2)
- KCNE2 (LQTS Typ 6 / acquired LQTS)
- KCNJ2 (Andersen-Tawil-Syndrom / LQTS Typ 7)
- CACNA1C (Timothy-Syndrom / LQTS Typ 8)
- CALM1 (LQT14), CALM2 (LQTS Typ 15) und CALM3 (LQTS Typ 16)
- TRDN
Die Kranksheitsrelevanz weiterer Gene (ANK2, AKAP9, CAV3, KCNE3, KCNJ5, SNTA1, SCN4B und TECRL), die vor allem historisch in Verbindung mit dem LQTS stehen, gilt heute als limitiert oder umstritten, sodass die humangenetische Analyse dieser Gene nicht mehr Teil der LQTS-Routinediagnostik ist. Auf Wunsch können diese Gene jedoch dennoch untersucht werden. Des Weiteren können Arzneistoffe verschiedenster Klassen eine Verlängerung der QT-Zeit hervorrufen. Ein verzögerter Metabolismus von Medikamenten, der durch Varianten im -, – oder -Gen bedingt sein kann, kann diesen Effekt verstärken. Beim medikamenten-induzierten Long QT-Syndrom kann die ergänzende Diagnostik der sinnvoll sein.