Kraniosynostose
Kraniosynostosen sind vorzeitige Verknöcherungen von Schädelnähten, die zu veränderten Kopfformen führen und in 1 von 2.000-3.000 Fällen auftreten. Sie können isoliert vorkommen oder als Teil komplexer Syndrome wie dem Apert-, Crouzon-, Muenke-, Pfeiffer- und Saethre-Chotzen-Syndrom. Die meisten dieser Syndrome werden autosomal-dominant vererbt und beruhen auf genetischen Veränderungen, insbesondere in den Fibroblasten-Wachstumsfaktor-Rezeptor-Genen (FGFR) oder im TWIST1-Gen.
Wissenschaftlicher Hintergrund
Als Kraniosynostose wird die vorzeitigeVerknöcherungvonSchädelnähten bezeichnet. Daraus ergibt sich in Abhängigkeit davon, welche Schädelnähte von der Synostose betroffen sind, ein verändertes Schädelwachstum mit z.T. charakteristischen Kopfformen. Die meisten primären Kraniosynostosen sind angeboren und können isoliert oder als Teilsymptom verschiedener komplexer Syndrome auftreten. Die Gesamthäufigkeit liegt bei 1:2.000-3.000. Neben exogenen Ursachen (z.B. extreme Frühgeburt, Valproat-Embryopathie u.a.) werden bei ca. 25% aller Kraniosynostosen monogene Ursachen gefunden.
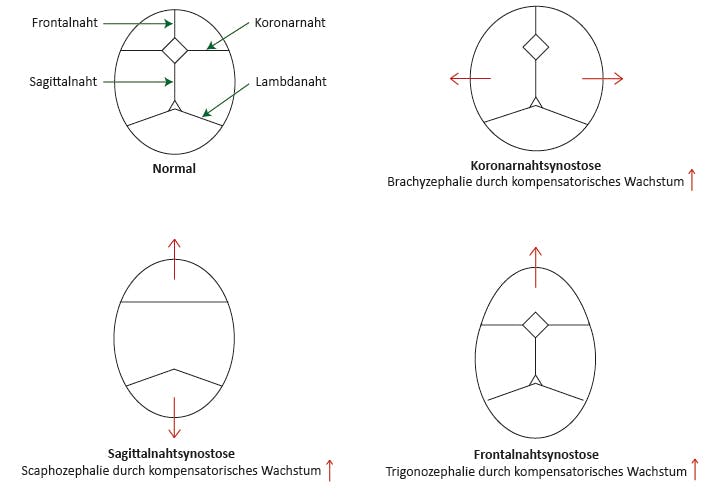
Abb.: Schematische Darstellung einiger Kraniosynostosen
Die komplexen Formen zeigen eine oder mehrere Synostosen und z.T. weitere Symptome einer pathologischen Entwicklung knöcherner Strukturen wie z. B. Syndaktylien. Zu den klassischen Kraniosynostosesyndromen zählen:
Diese Syndrome werden autosomal-dominant vererbt und beruhen mit Ausnahme des Saethre-Chotzen-Syndroms auf Veränderungenin den Genen der Fibroblasten-Wachstumsfaktor-Rezeptoren (FGFR) 1, 2 und 3. Das Saethre-Chotzen-Syndrom wird meist durch pathogene Veränderungen des TWIST1-Gens verursacht. Zwischen den Syndromen gibt es klinisch Überlappungen.
Unter den isolierten Formen sind die Sagittalnaht- bzw. Metopicasynostose mit einem Anteil von ca. 50% die häufigsten, sie haben aber nur in weniger als 1% nachweisbare genetische Ursachen, am ehesten Varianten in TWIST1. Bei einseitigen Koronarnahtsynostosen werden ursächliche Varianten in ca. 13% gefunden, bei beidseitigen Koronarnahtsynostosen in ca. 60%, bei Synostosen mehrerer Nähte in ca. 15%. Häufigste Ursache isolierter Koronarnahtsynostosen sind zur Haploinsuffizienz führende Varianten in TCF12.
Die molekulare Ursache der vorzeitigen Nahtverknöcherung ist noch nicht im Einzelnen verstanden. Die beteiligten Fibroblasten-Wachstumsfaktor-Rezeptoren sind Tyrosinkinase-Rezeptoren, die in der Ossifikation eine wichtige Rolle spielen. TWIST interagiert mit FGFR.
Komplikationen sind Erhöhung des intrakraniellen Drucks, Sehbeeinträchtigung und Hörstörungen und bei einigen der Syndrome auch eine Entwicklungsverzögerung. Die Therapie ist operativ.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.