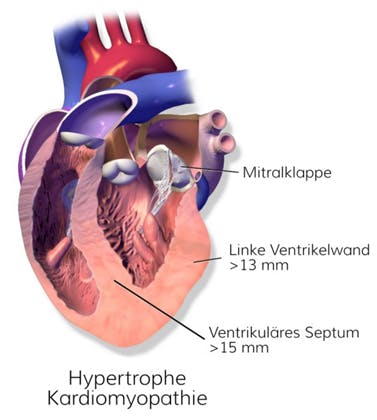
Hypertrophe Kardiomyopathie (HCM)
Die hypertrophe Kardiomyopathie (HCM) ist eine überwiegend autosomal‑dominant vererbte Erkrankung des Herzmuskels und durch eine Verdickung (Hypertrophie) des linken Ventrikels charakterisiert. Sie betrifft etwa eine von 500 Personen der Allgemeinbevölkerung und ist mit einem erhöhten Risiko für einen plötzlichen Herztod verbunden. Pathogene Varianten in über 100 Genen wurden beschrieben, wobei rund 90 Prozent der Befunde die Gene MYH7, MYBPC3, TNNT2 und TNNI3 betreffen, die zentrale strukturelle Proteine des Sarkomers kodieren.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Wissenschaftlicher Hintergrund
Erstellt von:
Die Medicover Genetics Fachredaktion besteht aus Fachärzt:innen und Wissenschaftler:innen im Bereich Humangenetik. Alle Inhalte werden nach aktuellen wissenschaftlichen Standards erstellt und geprüft.