Fragiles-X-Syndrom (FRAXA-Locus)
Das Fragile-X-Syndrom ist eine monogen vererbte Erkrankung, die durch eine CGG-Triplett-Repeat-Expansion im FMR1-Gen auf dem X-Chromosom verursacht wird. Es ist die häufigste monogen vererbte Ursache einer mentalen Retardierung. Normalerweise haben die meisten Menschen 29-30 CGG-Repeats, bei Patienten mit dem Fragilen-X-Syndrom erweitert sich diese Sequenz auf über 200 Repeats. Diese Expansion führt zur Methylierung von Cytosinresten des Repeats und benachbarter regulatorischer Elemente, was die Transkription des Gens hemmt und das FMR1-Genprodukt ausfallen lässt. Bei Frauen kann die Weitergabe einer Prämutation (55 bis 200 CGG-Repeats) an die nächste Generation zu einer Verlängerung auf über 200 Tripletts führen.
Wissenschaftlicher Hintergrund
Das Fragile-X-Syndrom ist die häufigste monogen vererbte Ursache einer mentalen Retardierung. Abweichend von anderen Erkrankungen mit X-chromosomal rezessivem Erbgang zeigt das Fragile-X-Syndrom Besonderheiten wie gesunde männliche Überträger und bei einem Teil der weiblichen Betroffenen eine ähnlich schwere Symptomatik wie im männlichen Geschlecht. Die Inzidenz betroffener männlicher Patienten wird nach einer Studie von 2009 mit 1:5164 beziffert.
Ursache des Fragilen-X-Syndroms ist eine CGG-Triplett-Repeat-Expansion im 5’-untranslatierten Bereich des FMR1-Gens (fragiles X-mentale Retardierung) auf dem langen Arm des X-Chromosoms. Die häufigsten Normalallele in der Allgemeinbevölkerung haben eine Länge von 29-30 CGG-Repeats. Allele im Bereich von 45 bis 54 Repeats werden als Grauzonenallel definiert (EMQN Ringversuch). Bei dieser Repeat-Anzahl liegt, unabhängig vom Geschlecht des Überträgers, bereits eine gewisse Instabilität vor, jedoch wurde in diesem Bereich noch keine Expansion zu einer Vollmutation in einer Generation beobachtet.
Als Prämutation werden Allele mit 55 bis 200 CGG-Repeats bezeichnet. Im weiblichen Geschlecht werden Prämutationen bei der Weitergabe an die nächste Generation instabil vererbt, was meist zu einer Verlängerung auf über 200 Tripletts (Vollmutation) führt. Ab dieser Länge kommt es zur Methylierung von Cytosinresten des Repeats und benachbarter regulatorischer Elemente, was letztlich zu einer Hemmung der Transkription und damit zum Ausfall des FMR1-Genprodukts führt. Im männlichen Geschlecht wird die Prämutation stabil an die nächste Generation weitergegeben. Mütter von Kindern mit Vollmutation sind obligate Überträgerinnen mit entweder einer Prämutation oder bereits einer Vollmutation. Das Wiederholungsrisiko beträgt bis zu 50% für betroffene Kinder in Abhängigkeit vom Geschlecht bzw. von der Länge der Prämutation bei der Mutter.
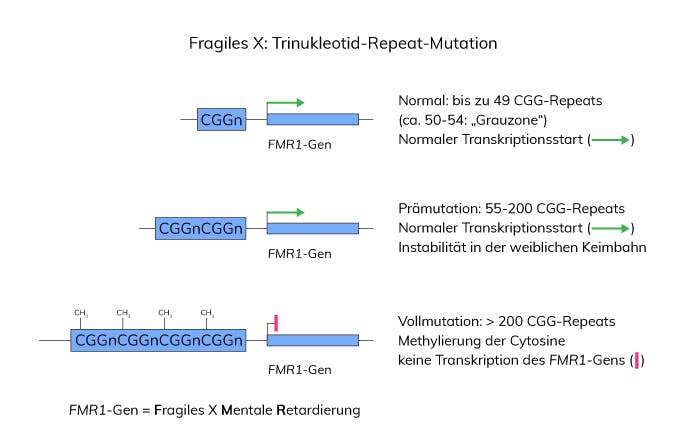
Abb.: Schematische Darstellung des FMR1-Gens mit Normalbefund, Prämutation und Vollmutation
Bei Vorliegen der Vollmutation fällt im Kindesalter zunächst die Entwicklungsverzögerung auf, die meist mehr die Sprache als die Motorik betrifft. Die Kinder haben oft etwas überdurchschnittliche Maße für Körperlänge und Kopfumfang. Gelegentlich fallen Symptome einer Bindegewebsschwäche wie überstreckbare Gelenke und Muskelhypotonie auf. Als charakteristische Merkmale werden Hyperaktivität und autistische Verhaltensweisen beobachtet. Abgesehen von eher großen Ohrmuscheln sind weitere Phänotypmerkmale wie längliches Gesicht und betontes Kinn im Kindesalter noch wenig ausgeprägt, kennzeichnen aber Erwachsene mit Fragilem-X-Syndrom. Postpubertär ist bei Männern häufig eine Makroorchie festzustellen. WeiblicheTrägerinnen der Vollmutation können eine sehr variable Symptomatik zeigen von einem unauffälligen Phänotyp (ca. 30%) bis zu einer ähnlich schweren mentalen Retardierung wie bei Männern. Etwa 20% der Prämutationsträgerinnen haben eine vorzeitige Menopause (FXPOI). Bei älteren, v.a. männlichen Prämutationsträgern zeigt sich zu über 30% ein oft progredientes neurologisches Krankheitsbild, bestehend aus Intentionstremor, Gangataxie, Parkinsonismus, autonomer Dysfunktion und Demenz, das mittlerweile als „Fragiles-X-Tremor-Ataxie- Syndrom“ (FXTAS) bezeichnet wird.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Indikation | ICD—10 | Gen | OMIM—G |
| Fragiles-X-Syndrom | Q99.2 | FMR1 | 309550 |