Fettstoffwechselstörungen
Fettstoffwechselstörungen gehören zu den Hauptrisikofaktoren für Herz-Kreislauf-Erkrankungen, die die häufigste Todesursache in Deutschland darstellen. Bei dieser Art von Störungen liegt eine Erhöhung beziehungsweise eine Verschiebung der Zusammensetzung der Lipide im Blut vor. Dies kann zu Arteriosklerose und in der Folge zu Herz-Kreislauf-Erkrankungen führen. Eine Fettstoffwechselstörung kann sowohl genetisch bedingt (primäre Fettstoffwechselstörung), als auch Folge anderer Grunderkrankungen wie z.B. Diabetes mellitus, Hypothyreose, Nierenerkrankungen oder anderer Stoffwechselerkrankungen sein (sekundäre Fettstoffwechselstörung). Die primären Fettstoffwechselstörungen werden unterteilt in , , , und . Die genetische Diagnostik dient der Diagnosesicherung und ist im Rahmen einer Familienuntersuchung relevant.
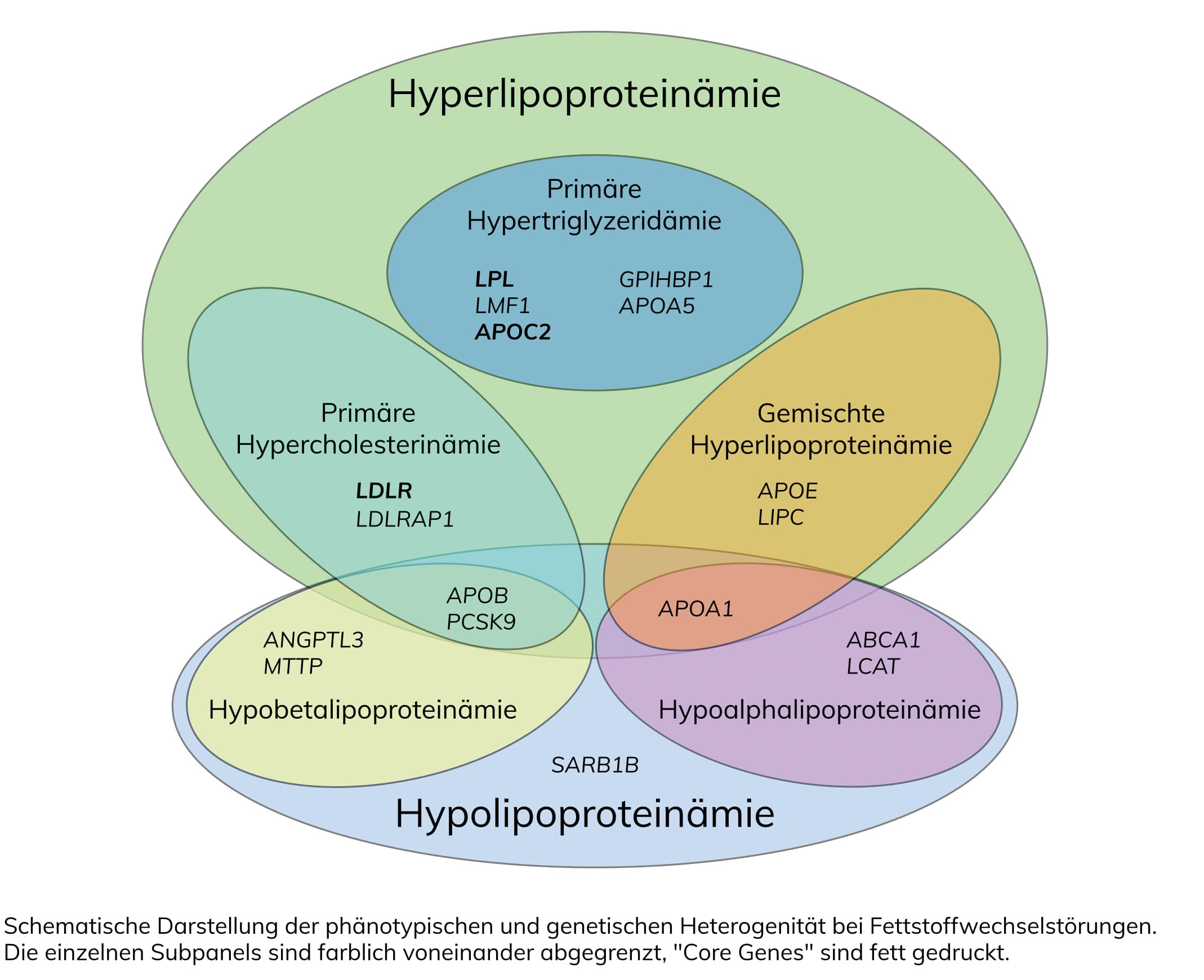
Detaillierte Informationen zu den einzelnen Erkrankungen finden Sie hier: