Dilatative Kardiomyopathie (DCM)
Die dilatative Kardiomyopathie (DCM) ist eine Erkrankung, bei der der linke oder beide Ventrikel des Herzens erweitert und ihre Kontraktionsfähigkeit beeinträchtigt sind. Obwohl bis zu 40% der idiopathischen Fälle genetisch bedingt sind, sind die genetischen Ursachen der DCM heterogen mit über 40 bekannten ursächlichen Genen. Insbesondere Varianten im Titin-Gen (TTN) stehen in etwa 25% der Fälle in ursächlichem Zusammenhang. Die frühe Identifikation von Analageträgern kann entscheidend sein, um den Krankheitsverlauf zu beeinflussen und ggfs. geeignete Therapien zu wählen.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Wissenschaftlicher Hintergrund
Die dilatative Kardiomyopathie (DCM) ist durch die Dilatation und eingeschränkte Kontraktion des linken oder beider Ventrikel charakterisiert. Die Prävalenz liegt bei ungefähr 1:2.500. Meist geht die Erkrankung mit einer progredienten Herzinsuffizienz einher. Es besteht ein deutlich erhöhtes Risiko für Arrhythmien, Thromboembolien und plötzlichen Herztod. Obwohl die Therapie sich verbessert hat, wurden 5-Jahres-Überlebensraten von 36-80% ermittelt. Es konnte gezeigt werden, dass nur die Hälfte der DCM-Fälle mit scheinbar unklarer Ursache tatsächlich idiopathisch sind (IDCM) und nicht sekundär auf anderen Primärerkrankungen basieren. Folglich ist es wichtig, vor der Veranlassung einer genetischen Diagnostik eine sekundäre dilatative Kardiomyopathie möglichst vollständig auszuschließen. In bis zu 40% der Fälle ist die IDCM genetisch bedingt und meist autosomal-dominant vererbt. Die frühe Identifikation von Anlageträgern ist essentiell, um den Verlauf der Erkrankung positiv zu beeinflussen. Der Phänotyp hängt auch von der Penetranz und der Malignität des betroffenen Gens ab. Patienten mit pathogenen TTN-Varianten zeigen meist einen milderen Verlauf und sprechen besser auf Therapie an. Allerdings wurden bei diesen auch häufiger ventrikuläre Arrhythmien beobachtet. Patienten mit pathogenen Varianten in den Genen LMNA, PLN, RBM20, FLNC, DES und SCN5A haben ein erhöhtes Risiko für eine schlechtere Prognose und für maligne Arrhythmien.
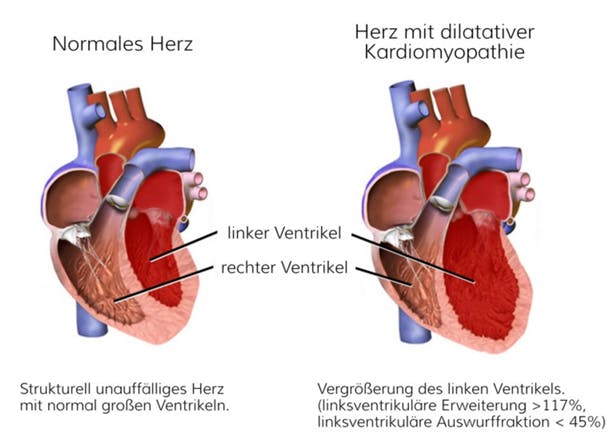
Abb.: Darstellung der strukturellen Veränderung bei einem Herz mit dilatativer Kardiomyopathie (mod. nach BruceBlaus (own work) [CC BY-SA 4.0], via Wikimedia Commons)
Die genetischen Ursachen der IDCM/FDCM sind heterogen; inzwischen sind über 40 ursächliche Gene bekannt. Vor über zehn Jahren wurden bereits Varianten im LMNA-Gen, das für Strukturproteine der inneren Zellkernmembran codiert, im Zusammenhang mit DCM identifiziert. Eine große Studie ergab, dass ca. 6-8% der IDCM/FDCM-Patienten Varianten in LMNA tragen. Gehäuft wurden außerdem Veränderungen in verschiedenen Sarkomer-Proteinen als Ursache der IDCM/FDCM nachgewiesen. Ungefähr ein Viertel aller Fälle tragen Varianten in den Genen der schweren Kette des ß-Myosins (MYH7), des Myosinbindeproteins-C (MYBPC3) und des Troponins T (TNNT2). Varianten in diesen Genen wurden zwar wesentlich häufiger im Zusammenhang mit HCM beschrieben, inzwischen sind allerdings auch über 100 verschiedene, für die dilatative Kardiomyopathie spezifische Varianten bekannt. Mit der Analyse von vier gehäuft betroffenen Genen können in ungefähr einem Drittel aller IDCM/FDCM-Fälle ursächliche Varianten nachgewiesen werden.
In neueren Studien konnte an über 300 IDCM-Patienten gezeigt werden, dass Varianten im größten menschlichen Gen Titin (TTN) in ca. 25% der Fälle in ursächlichem Zusammenhang stehen. Hierbei handelt es sich um schwerwiegende Varianten, die zum funktionellen Verlust führen. Die Penetranz dieser Varianten ist jedoch nicht vollständig, sodass die Ursächlichkeit in jeder Familie durch die gezielte Analyse mehrerer Familienmitglieder abgesichert werden sollte. Dieses Gen kann aufgrund der Größe nur mittels neuer Sequenzierverfahren analysiert werden. Dieses Verfahren bietet zudem den Vorteil, dass über 30 Gene, in denen gehäuft Varianten im Zusammenhang mit DCM nachgewiesen wurden, parallel analysiert werden können.