Smith-Lemli-Opitz-Syndrom
Das rezessiv vererbte Smith-Lemli-Opitz-Syndrom (SLOS) ist eine Störung im Syntheseweg des Cholesterins. Ursächlich sind pathogene Varianten im DHCR7-Gen, die zum Verlust der Enzymaktivität der 7-Dehydrocholesterin-Reduktase führen und somit zum Ausfall des letzten Schrittes in der Cholesterinsynthese. Die Anreicherung von 7-Dehydrocholesterin (und seinem Isomer 8-Dehydrocholesterin) führt zum Erkrankungsbild.
Wissenschaftlicher Hintergrund
Das Smith-Lemli-Opitz-Syndrom (SLOS) ist je nach Schweregrad gekennzeichnet durch eine breit gefächerte Symptomatik mit Dysmorphiezeichen (Mikrozephalie, „Mittelliniendefekt“, Mikroretrognathie, tiefsitzende Ohren, Blepharoptose, postaxiale Polydaktylie), psychomotorischer Retardierung, Minderwuchs sowie Fehlbildungen wie Herzfehler und (LK)G-Spalten. Betroffene Jungen können darüber hinaus durch Genitalfehlbildungen auffallen (z.B. Hypospadie). Die molekulare Ursache findet sich in einer Störung des letzten Schrittes im Syntheseweg des Cholesterins,wodurch es zur Anreicherung von 7-Dehydrocholesterin (und seinem Isomer 8-Dehydrocholesterin) kommt. Da die herkömmliche enzymatische Cholesterinbestimmung Cholesterin nicht von 7-Dehydrocholesterin differenzieren kann, erfolgt der biochemische Nachweis des SLOS über Gaschromatographie/Massenspektrometrie. Ein erhöhtes Dehydrocholesterin/Cholesterin-Verhältnis ist dabei hinweisgebend für das SLOS.
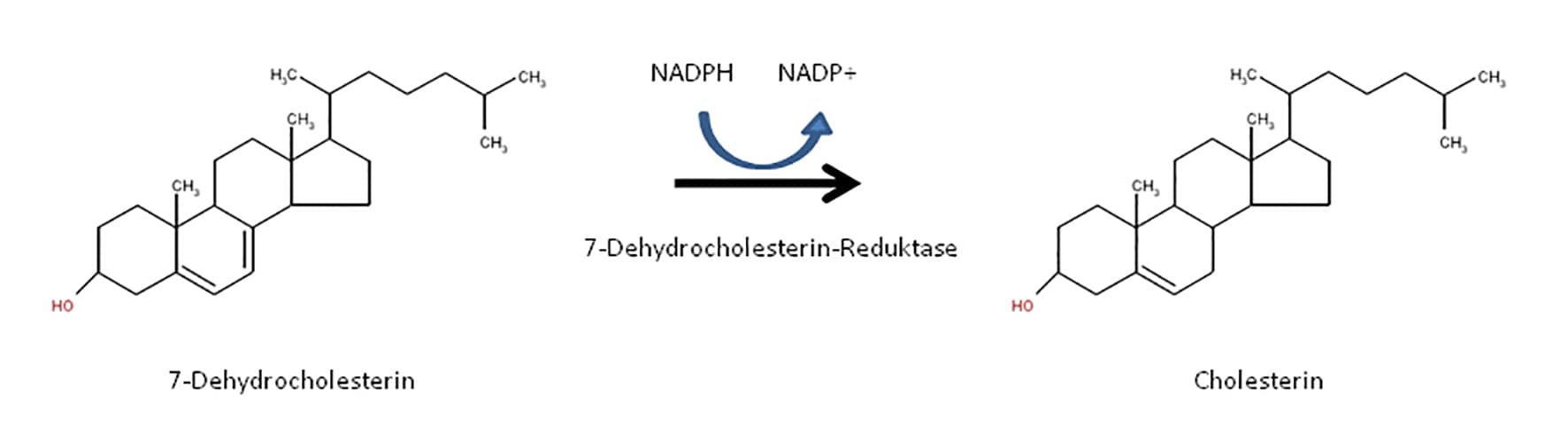
Die Vererbung des SLOS folgt einem autosomal rezessiven Erbgang. Die Prävalenz liegt bei 1:10.000 bis 1:30.000. Pathogene Varianten im DHCR7-Gen, das für das Enzym 7-Dehydrocholesterin-Reduktase codiert, führen zum Verlust der Enzymaktivität und somit zum Ausfall des letzten Schrittes in der Cholesterinsynthese. Die häufigste ursächliche Variante (c.964-1G>C) liegt in Intron 8 des DHCR7-Gens. Sie lässt sich bei ca. 30% aller Patienten nachweisen. Art der Variante und Locus im Gen beeinflussen den Schweregrad des SLOS. Eine eindeutige Genotyp-Phänotyp-Korrelation ist jedoch nicht immer gegeben, so dass Patienten mit den gleichen Varianten unterschiedlich stark betroffen sein können. Daher werden weitere Faktoren diskutiert, die Einfluss auf den Phänotyp haben. In einer Studie von 2004 konnte ein Zusammenhang zwischen maternalem und dem Schweregrad bei Patienten mit SLOS nachgewiesen werden.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
Indikation | ICD—10 | Gen | OMIM—G |
| Smith-Lemli-Opitz-Syndrom | Q87.1 | DHCR7 | - |