Long QT-Syndrom (LQTS)
Das Long-QT-Syndrom (LQTS) ist eine häufig genetisch bedingte Herzerkrankung, die durch eine verlängerte ventrikuläre Repolarisation und ein erhöhtes Risiko für lebensbedrohliche Arrhythmien gekennzeichnet ist. Pathogene Varianten in den Genen KCNQ1, KCNH2 und SCN5A machen etwa 95 Prozent der genetisch gesicherten Fälle aus.
* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.
- Long QT-Syndrom (LQTS)19 GeneAKAP9ANK2CACNA1CCALM1CALM2CALM3CAV3KCNE1KCNE2KCNE3KCNH2KCNJ2KCNJ5KCNQ1SCN4BSCN5ASNTA1TECRLTRDN
Zur Anforderung der erweiterten Diagnostik* bitte den verwenden.
*erweitert um Gene mit derzeit noch nicht hinreichend geklärter Krankheitsrelevanz (Gene unklarer Signifikanz).
Wissenschaftlicher Hintergrund
Erstellt von:
Die Medicover Genetics Fachredaktion besteht aus Fachärzt:innen und Wissenschaftler:innen im Bereich Humangenetik. Alle Inhalte werden nach aktuellen wissenschaftlichen Standards erstellt und geprüft.
Das Long QT-Syndrom (LQTS) ist eine klinisch und genetisch heterogene Herzerkrankung, die durch eine verlängerte ventrikuläre Repolarisation charakterisiert ist. Typisch sind frequenzkorrigierte QT-Intervalle (QTc) von etwa 460 bis über 500 Millisekunden. Auf molekularer Ebene beruht die QT-Verlängerung auf einer Reduktion auswärtsgerichteter Kaliumströme, vor allem IKs, IKr und IK1, oder auf einer Zunahme einwärtsgerichteter Ströme, insbesondere INa und ICaL. Mit zunehmender Verlängerung des QTc‑Intervalls steigt das Risiko für ventrikuläre Arrhythmien deutlich an, was sich klinisch als Synkope oder als potenziell tödliches Rhythmusereignis manifestieren kann. Die Zehnjahresmortalität beträgt unbehandelt etwa 50 Prozent.
Es wird zwischen der häufigen autosomal-dominanten Romano-Ward-Form und der seltenen autosomal-rezessiven Jervell-Lange-Nielsen-Form unterschieden, die biallelische pathogene Varianten in KCNQ1 oder KCNE1 mit ausgeprägten QTc-Verlängerungen und kongenitaler sensorineuraler Hörstörung kombiniert. Aktuelle Daten zeigen jedoch, dass die klinischen Ausprägungen abhängig von Art und Kombination der Varianten ein breites Spektrum bilden. Heterozygote Träger sehr milder oder gering penetranter Varianten zeigen häufig keine oder nur minimale klinische Auffälligkeiten. Liegen diese milderen Varianten jedoch homozygot oder in kombiniert heterozygoter Form vor, kann sich ein Long‑QT‑Phänotyp ohne Hörverlust entwickeln, der dem Romano‑Ward‑Bild ähnelt.
Die Prävalenz des LQTS beträgt etwa 1:2000 – 1:2500.
Die Diagnosestellung sollte nicht allein auf einem QTc-Grenzwert beruhen, sondern die EKG-Befunde, klinische Symptome, die Familienanamnese und genetische Ergebnisse einbeziehen. Ein QTc von mindestens 480 ms in wiederholten Ableitungen ohne sekundäre Ursachen, ein ausreichend hoher Schwartz-Score oder eine nachgewiesene pathogene LQTS-assoziierte Variante können jeweils die Diagnose sichern. QTc-Werte im Grenzbereich ab etwa 460 Millisekunden unterstützen die Diagnose insbesondere bei Patienten mit ungeklärten Synkopen oder überlebtem plötzlichen Herztod.
Pathogene Varianten in KCNQ1, KCNH2 und SCN5A werden in etwa 90 bis 95 Prozent der molekulargenetisch gesicherten LQTS-Fälle identifiziert. KCNQ1 und KCNH2 kodieren die alpha-Untereinheiten der kardialen Kaliumkanäle Kv7.1 und Kv11.1, welche den IKs- sowie den IKr-Strom vermitteln. Beide Ströme sind für die Repolarisation des kardialen Aktionspotenzials essentiell. Das SCN5A-Gen kodiert den Natriumkanal Nav1.5, der den Natriumeinstrom INa vermittelt und an der schnellen Depolarisation beteiligt ist. Eine pathologisch reduzierter Kaliumstrom oder ein pathologisch erhöhter Natriumstrom bzw. ein Natrium-Leckstrom kann die Dauer der ventrikulären Repolarisation und damit das QTc-Intervall verlängern.
Die molekulare Klassifikation und Nomenklatur orientiert sich hierbei an den betroffenen Genen:
- KCNQ1 (LQTS Typ1; 45 % der pathogenen Varianten, eigene Daten) Varianten mit dominanter oder rezessiver Ausprägung (RW- und JLN-Form) sind beschrieben. Patienten mit pathogenen Varianten im KCNQ1-Gen zeigen meist frühzeitig einen deutlich ausgeprägten Phänotyp mit einem hohen Risiko für kardiale Ereignisse.

Abb.: LQTS1 EKG mit verlängerter QT-Zeit (rote Pfeile)
- KCNH2 (LQTS Typ2; 39 % der pathogenen Varianten, eigene Daten) Es besteht ein hohes Risiko für kardiale Ereignisse. Es handelt es sich um RW-Formen.
- SCN5A (LQTS Typ3; 8 % der pathogenen Varianten, eigene Daten) Kardiale Ereignisse sind seltener, die Letalität ist jedoch fünffach höher. Klinisch tritt LQTS Typ 3 als RW-Form auf.
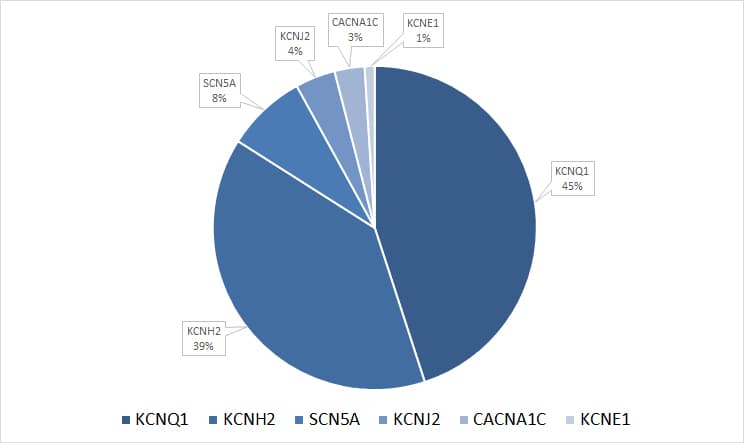
Abb.: Verteilung gefundender, ursächlicher Varianten auf sechs myokardiale Ionenkanal-Gene, in welchen zu 75% aller klinisch gesicherten LQTS-Fälle pathogene Veränderungen nachweisbar sind (eigene Daten).
Seltene Formen des LQTS können durch pathogene Varianten in weiteren Genen wie CACNA1C, CALM1, CALM2, CALM3, KCNE1, KCNE2, KCNJ2 oder TRDN verursacht sein. Einige dieser Gene sind mit komplexen oder syndromalen Erkrankungen verknüpft, etwa das KCNJ2-assoziierte Andersen-Tawil-Syndrom, welches eine QTs-Verlängerung und eine periodische Hyperkaliämie kombiniert. Pathogene Varianten in den CALM-Genen können zu schweren, früh manifestierenden arrhythmogenen Erkrankungen führen, die Merkmale eines LQTS und einer katecholaminergen polymorphen ventrikulären Tachykardie zeigen. Des Weiteren können Arzneistoffe verschiedenster Klassen eine Verlängerung der QT-Zeit hervorrufen. Ein verzögerter Metabolismus von Medikamenten, der durch Varianten im -, – oder -Gen bedingt sein kann, kann diesen Effekt verstärken. Beim medikamenten-induzierten Long QT-Syndrom kann die ergänzende Diagnostik der sinnvoll sein.
Die Identifikation von Trägern pathogener Varianten ermöglicht frühzeitige und potenziell präsymptomatische Therapieentscheidungen. Das Risiko kardialer Ereignisse sinkt um etwa 62 bis 95 Prozent für das LQTS Typ 1 und um etwa 74 Prozent für das LQTS Typ 2. Es wird empfohlen, die neuesten ESC-Leitlinien bezüglich des Managements von hereditären Arrhythmien und der Prävention des plötzlichen Herztodes zu Rate zu ziehen. Sie empfehlen (Klasse I) allen LQTS-Patienten ab dem Zeitpunkt der Diagnose den Lebensstil anzupassen (z. B. die Vermeidung von QT-verlängernden Arzneimitteln, siehe crediblemeds.org) und mit ß-Blockern behandelt zu werden. Die Implantation eines ICDs wird bei therapierefraktären rezidivierenden Synkopen / ventrikulären Tachykardien oder einem vorangegangenen Herzstillstand empfohlen (Klasse I). Der Risikorechner der Universität Rochester ermöglicht die Prognose des absoluten 5-Jahres-Risikos für das erste lebensbedrohliche Arrhythmieereignis bei LQTS-Patienten ().
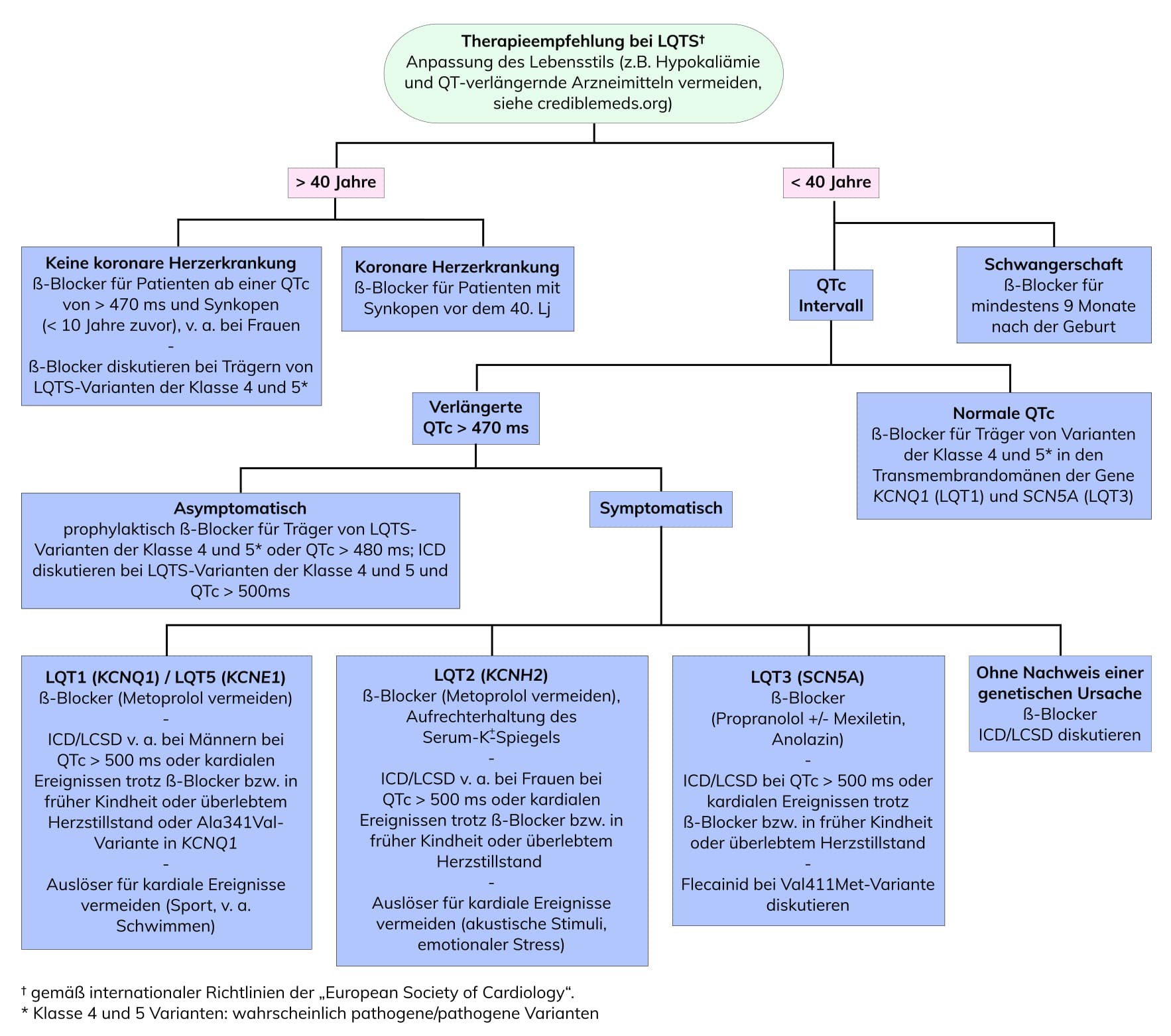
Abb.: Therapieempfehlung bei Long QT-Syndrom, modifiziert nach Adamos et al. 2018, Mini Rev Med Chem 18:495 / Giudicessi et al. 2013, Curr Probl Cardiol 38:417 / Priori et al. 2015, European Heart Journal 36:2793
Crotti et al. 2023, Eur Heart J 44:3357 / Zeppenfeld et al. 2022, European Heart Journal 43:3997 / Adler et al. 2020, Circulation 141:418 / Garcia-Elias et al. 2018, Int J Mol Sci 19:692 / Giudicessi et al. 2018, Trends Cardiovasc Med 28:453 / Itoh et al. 2016, Eur J Hum Genet 24:1160 / Lieve et al. 2013, Genet Test Mol Biomarkers 17:553 / Ackerman et al. 2011, Europace 13:1077 / Tester et al. 2010, Am J Cardiol 106:1124
Häufig gestellte Fragen
Vorwiegend werden pathogene Varianten in KCNQ1, KCNH2 und SCN5A identifiziert. Die Untersuchung weiterer Gene wie KCNE1, KCNE2, KCNJ2, CACNA1C, CALM1, CALM2, CALM3 oder TRDN sollte integraler Bestandteil eines umfassenden LQTS-Panels sein, da sie gemeinsam zu einer vollständigen genetischen Abklärung beitragen. Auch wenn pathogene Varianten in diesen weiteren Genen bei weniger als fünf Prozent der LQTS‑Patienten nachgewiesen werden, sind sie klinisch dennoch bedeutsam, da sie mit spezifischen und zum Teil syndromalen oder schweren arrhythmogenen Phänotypen verbunden sein können.
Genetische Untersuchungen sind besonders hilfreich bei Patienten, deren klinische Befunde oder Familienanamnese den Verdacht auf ein LQTS nahelegen, einschließlich ungeklärter Synkopen, plötzlicher Todesfälle in der Familie oder charakteristischer EKG-Auffälligkeiten. Auch bei zufällig festgestellten grenzwertigen oder mild verlängerten QTc-Werten kann eine genetische Abklärung indiziert sein, da die Penetranz und Ausprägung des LQTS stark variieren und Träger nur diskrete oder subklinische Merkmale zeigen können.
Der Nachweis einer Variante ermöglicht rechtzeitige und gegebenenfalls präsymptomatische therapeutische Maßnahmen wie das Vermeiden bestimmter Medikamente und die Einnahme von Betablockern. Die Identifikation einer ursächlichen Variante unterstützt die Subtypisierung, erleichtert die Risikostratifizierung, fördert eine gezielte Überwachung und therapeutische Entscheidungen und ist für die prädiktive Diagnostik bei Angehörigen von entscheidender Bedeutung.