Alzheimer Erkrankung - Frühform (AD1)
Die Alzheimer-Krankheit ist die häufigste Form der Altersdemenz, wobei genetische Faktoren eine Rolle spielen. Varianten in den Genen Präsenilin 1 (PSEN1), Präsenilin 2 (PSEN2) und Amyloid-Vorläufer-Protein (APP) führen zur früh einsetzenden, autosomal-dominant vererbten Form von Alzheimer. Diese Gene sind an der Prozessierung des Beta-Amyloid-Vorläufer-Proteins zu Beta-Amyloid beteiligt, das ein wesentlicher Bestandteil der neuritischen Plaques ist. In Familien mit Betroffenen in mindestens drei Generationen sind etwa 60% der Fälle auf Varianten im PSEN1-Gen und ca. 20% auf Varianten im APP-Gen zurückzuführen.
Wissenschaftlicher Hintergrund
Die Alzheimer Erkrankung ist mit einer Prävalenz von 1:5 bei über 80-jährigen die häufigste Form der Altersdemenz. Für alle Formen der Alzheimer Erkrankung ist eine Beteiligung genetischer Faktoren bekannt.
Varianten im Präsenilin 1- (PSEN1), Präsenilin 2- (PSEN2), und im Amyloid-Vorläufer-Protein-Gen (APP) führen zur autosomal-dominant vererbten hereditären Frühform von M. Alzheimer (Beginn der Erkrankung vor dem 60. Lebensjahr). In Familien mit Betroffenen in mindestens drei Generationen sind etwa 60% der Fälle auf Varianten im PSEN1-Gen und ca. 20% auf Varianten im APP-Gen zurückzuführen. Eine weitere Ursache für die Erkrankung sind in <5% der Fälle Varianten im PSEN2-Gen. Für die verbleibenden ca. 15% gibt es zum Teil Hinweise auf Assoziation zu noch nicht näher charakterisierten Genen. PSEN1 und PSEN2 codieren für zwei der vier Proteine , die Bestandteil des Gamma-Sekretase-Proteinkomplexes sind. Dieser Komplex ist an der Prozessierung des Beta-Amyloid-Vorläufer- Proteins zu Beta-Amyloid beteiligt. APP codiert für das Beta-Amyloid-Vorläufer-Protein, ein ubiquitär exprimiertes, transmembranöses Protein. Varianten im PSEN1– oder APP-Gen führen zu einer Zunahme von Beta-Amyloid 42 (Aß42) zu Lasten von Aß40. Beide Proteine sind ein wesentlicher Bestandteil der neuritischen Plaques. Allerdings aggregiert das stärker hydrophobe Aß42 rascher zu den toxischen Fibrillen.
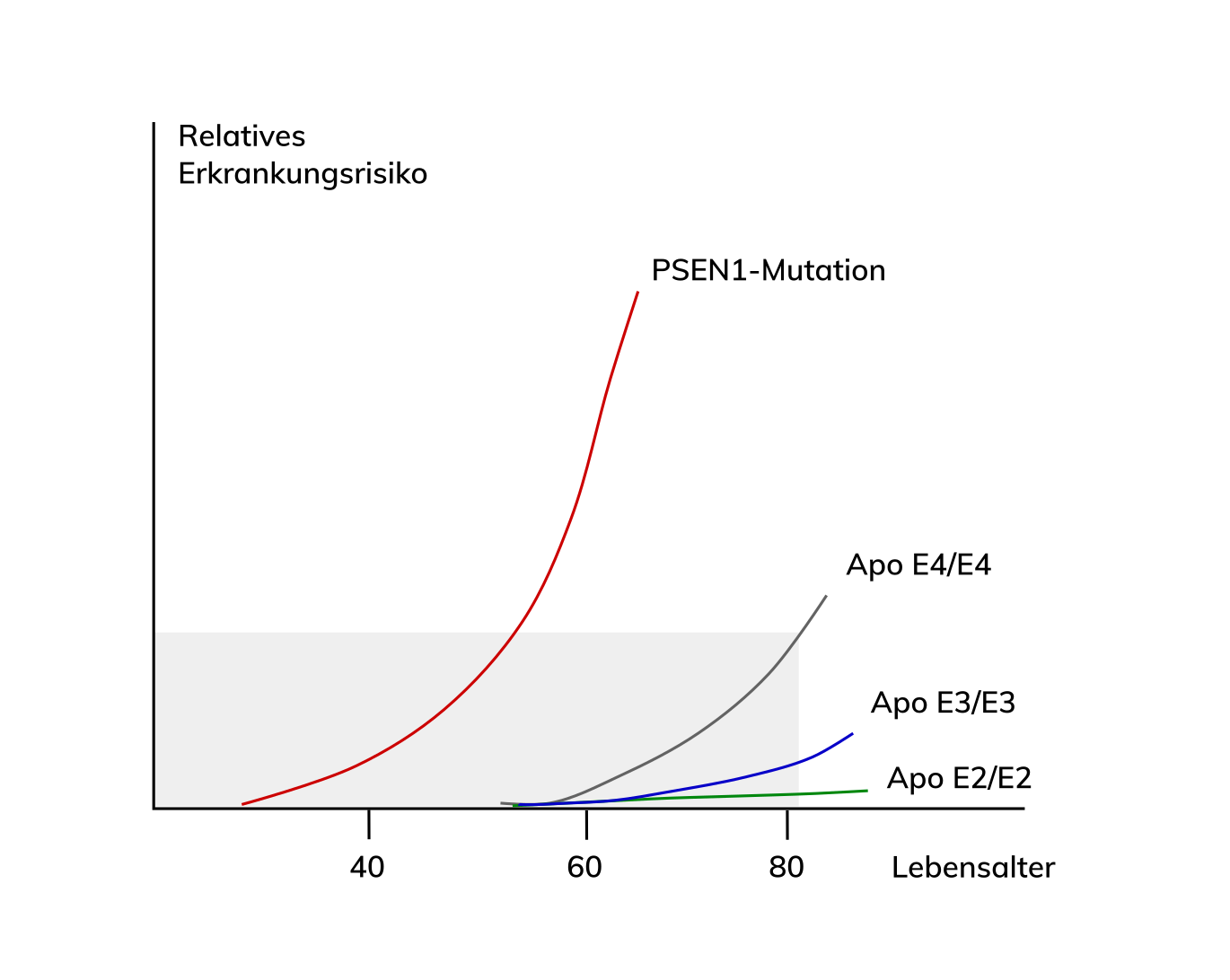
Detaillierte Informationen zur Assoziation der Alzheimer Erkrankung mit dem ApoE-Genotyp finden Sie auf unserer Seite .

* Aufgrund laufender Aktualisierungen können die hier aufgeführten Gene vorübergehend vom derzeit aktuellen Portfolio abweichen.